










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkRevista Uruguaya de Cardiología
On-line version ISSN 1688-0420
Rev.Urug.Cardiol. vol.26 no.1 Montevideo Mar. 2011
REGISTRO URUGUAYO
Previniendo el infarto en el adulto joven: GENYCO, un registro nacional de hipercolesterolemia familiar
En memoria del Profesor Orestes Fiandra, creador e inspirador
Dres. Mario Stoll 1, Mariana Lorenzo 1, Víctor Raggio 1, Patricia Esperón 1, Mario Zelarayan 2
1. Área de Genética Cardiovascular de la Comisión Honoraria para la Salud Cardiovascular.
2. Director Ejecutivo de la Comisión Honoraria para la Salud Cardiovascular.
Correspondencia: Dr. Mario Stoll. CHSCV. Br. Artigas 2358, Montevideo, Uruguay. Correo electrónico: mstoll@montevideo.com.uy
Recibido abril 26, 2011; aceptado mayo 6, 2011
RESUMEN
La detección de la hipercolesterolemia familiar permite identificar a un grupo poblacional con elevado riesgo genético utilizando la caracterización molecular de mutaciones en el gene del receptor de LDL, detección que es punto de partida para prevenir la enfermedad coronaria temprana. Los cardiólogos deben reconocer a estos pacientes y a sus grupos familiares, puesto que ello constituye una oportunidad de intervención precoz e individualizada. El programa GENYCO (GENes Y COlesterol) intenta revertir la situación de subdiagnóstico y subtratamiento de esta afección en nuestro medio, la que concierne fundamentalmente al adulto joven, existiendo entre 5.000 y 7.000 portadores de la misma. El tratamiento farmacológico con estatinas y otros hipolipemiantes reduce el riesgo de enfermedad coronaria prematura y pospone la cirugía coronaria en 10 a 20 años, consiguiéndose una sobrevida similar al resto de la población. En un programa piloto hemos identificado 245 afectados vivos con esta condición a partir de 71 casos índice, 18% de los cuales es portador de enfermedad coronaria, estando 85% de los hipercolesterolémicos familiares sin diagnóstico ni tratamiento. Esto implica un promedio de 3,1 afectados detectados a partir de cada caso índice por el método de seguimiento familiar en cascada. Se realizó el test molecular en 23 grupos familiares, identificándose 69 portadores de mutaciones en LDLR y 2 en ApoB100, realizándose el asesoramiento genético y médico. Mediante esta iniciativa pretendemos mejorar la detección y el control de estos individuos para de esa forma reducir la enfermedad coronaria precoz.
PALABRAS CLAVE:
HIPERCOLESTEROLEMIA
PREVENCIÓN PRIMARIA
GENÉTICA
GENÓMICA
DISLIPIDEMIAS
AGENTES HIPOLIPEMIANTES
SUMMARY
Familial hypercholesterolemia has been promoted internationally as an excellent model to demonstrate the benefit of prevention of coronary heart disease and early miocardial infarction in young adults, easily identifying a population group with high genetic risk, and using the molecular characterization of mutations in LDL receptor gene. Cardiologists in particular, should recognize these patients and family groups, and use the opportunity for an earlier and more individualized intervention. The GENYCO program (for Genes and Cholesterol) is intended to reverse the situation of insufficient diagnosis and inadequate treatment of a chronic disease that attacks young adults. In Uruguay we estimate between 5.000 and 7.000 FH affected patients. Pharmacological treatment of hypercholesterolemia with statins and other lipid-lowering drugs, based on knowledge of the molecular basis of this disease, produces a substantial reduction in risk of premature cardiovascular disease, postponed coronary artery bypass surgery 10 to 20 years and allows to achieve similar survival to the rest of the population. The pilot program, developed by the authors, identified 245 affected from 71 index cases with 18% of established coronary disease, 85% undiagnosed and untreated. This gives an average of 3.1 detections per index case by the family based cascade method. Molecular test was performed in 23 family groups, and identified 67 LDLR gene mutation carriers and 2 in ApoB100. We performed genetic and medical counseling. The tragedy of the death of young adults with coronary artery disease is reasonably preventable and avoidable.
Key words:
HYPERCHOLESTEROLEMIA
PRIMARY PREVENTION
GENETICS
GENOMICS
DYSLIPIDEMIAS
HYPOLIPIDEMIC AGENTS
INTRODUCCIóN
La hipercolesterolemia autosómica dominante (ADH del inglés: autosomal dominant hypercholesterolemia) que referimos comúnmente como hipercolesterolemia familiar (HF) o hipercolesterolemia familiar heterocigota (HFh) por su presentación más frecuente, se caracteriza clínicamente por una elevación constitutiva, desde el nacimiento, de los niveles plasmáticos de cLDL, y una aceleración en la progresión de la aterosclerosis a eventos cardiovasculares tempranos, provocando muerte cardiovascular en individuos a partir de los 30 años. El diagnóstico temprano de la HF es de enorme importancia ya que las terapias con fármacos hipolipemiantes han demostrado extensamente una franca disminución del riesgo de eventos (1).
El trabajo de Goldstein y Brown (premios Nobel de Fisiología y Medicina, 1985) en la HF y su descubrimiento de las mutaciones del receptor de LDL, suministró las bases para entender los mecanismos de la síntesis y el ciclo celular del colesterol, y para la búsqueda y el diseño de los inhibidores de la 5 hidroxi-3-metilglutaril-CoA reductasa (HMG-CoA), que resultaron los fármacos de mayor impacto en la protección cardiovascular en las dislipemias (2,3). Lamentablemente, los pacientes con HF quedaron relegados en el interés sanitario frente a dislipemias más comunes y la incidencia de otros factores de riesgo de la enfermedad coronaria, y expuestos al subdiagnóstico y subtratamiento. Analizaremos esta dislipemia genética, su clínica, sus mecanismos moleculares y sus consecuencias epidemiológicas, que nos han estimulado a conocer la situación en nuestra población y trabajar para su diagnóstico y prevención cardiovascular en las familias afectadas. La creación de GENYCO (GENes Y COLesterol) es el producto de ese trabajo.
EPIDEMIOLOGíA
Sabemos que la enfermedad cardiovascular (EC) tiene un tremendo impacto en la sociedad uruguaya en términos de mortalidad, morbilidad y en los costos sanitarios asociados. En conjunto la EC es la principal causa de muerte en Uruguay, América y Europa y lo será en todos los países del mundo para el año 2020. En nuestro país representa el 30% del total de muertes. En números absolutos, según la última encuesta de la Comisión Honoraria para la Salud Cardiovascular (CHSCV) (2008), ocurren unas 9.500 muertes por año, lo que da un promedio de 27 fallecimientos por día por esta causa. Cinco de esas muertes ocurren en menores de 65 años y cada dos días muere un uruguayo con menos de 45 años por EC.
Los promedios de los últimos años muestran que en Uruguay, en edades comprendidas entre los 30 y los 60 años, fallecen por causa de EC más de 80 individuos por mes. Paralelamente, se realizan más de 85 cateterismos mensuales dentro de este rango etario, que representa las tres décadas más productivas de la vida humana y donde la muerte tiene el mayor impacto familiar, social y económico.
Las dislipemias genéticas en general y la HF en particular ocupan el lugar más importante entre estas muertes en adultos jóvenes. Aunque Uruguay tiene una tasa promedio de mortalidad cardiovascular descendente desde hace tres décadas, el descenso es fundamentalmente a expensas de los mayores de 70 años, pero en los grupos etarios más jóvenes no existe un descenso tan marcado. El retraso en el diagnóstico de la HF, el subdiagnóstico y el subtratamiento son las causas mejor conocidas que inciden actualmente en la lentitud de este descenso.
La HF es una de las enfermedades hereditarias dominantes más comunes en todas las poblaciones conocidas, con una prevalencia de 1/400-1/500, afectando por igual a varones y mujeres. Existen poblaciones como los cristianos libaneses, los franco-canadienses o los afrikáners de la colonización holandesa en Sudáfrica, en las que un efecto fundador (mutación fundadora) ha aumentado la prevalencia hasta 1 en 100 individuos con mutaciones específicas. Sin embargo, en general las mutaciones son propias y específicas de cada familia y se han descrito más de 1.000 solo en el gene del receptor de LDL. Con una penetrancia de casi 100%, la presencia del gene mutado en dosis simple (heterocigosis) genera el fenotipo característico. La herencia determina, por lo tanto, un patrón de tipo dominante en donde el 50% de la descendencia de una persona afectada puede presentar el trastorno. Se estima que en Uruguay, considerando la frecuencia de la enfermedad, pueden existir entre 5.000 y 7.000 portadores de HF, lo que hace de esta población uno de los grupos de riesgo más importante, demandante de objetivos de prevención y tratamiento.
GENéTICA
El diagnóstico molecular de las dislipemias dominantes está basado fundamentalmente en el gene del receptor de LDL (RLDL) y las mutaciones frecuentes del gene de apolipoproteína B, (ApoB); ambas generan más de 98% de las dislipemias dominantes de alta frecuencia: la HF y la hipercolesterolemia familiar por defecto de ApoB. Ambos genes están directamente relacionados al transporte y reconocimiento de la partícula de LDL (tabla 1).

La enfermedad se produce con más frecuencia por una mutación del gene del receptor de las lipoproteínas de baja densidad (RLDL), situado en la zona distal del brazo corto del cromosoma 19. Este gene codifica un receptor transmembrana que capta e internaliza las partículas de LDL hacia el interior de las células hepáticas donde se metaboliza, retirándolas del torrente sanguíneo. Pertenece a una gran familia de receptores de membrana emparentados, que evolucionaron por duplicación y agregado de dominios con funciones diversas, pero fundamentalmente como interacción entre la membrana y el endosoma. La consecuencia de una mutación es una proteína defectuosa o que no se sintetiza, y, por tanto, una reducción importante en el número de receptores funcionales para las LDL a nivel hepático. Esto produce un aumento en las concentraciones plasmáticas de colesterol transportado en las lipoproteínas de baja densidad (cLDL), que supera los mecanismos de limpieza periféricos. La infiltración de colesterol en la pared de las arterias genera placas de ateroma que ocluyen la luz arterial y ambienta la trombosis y otros fenómenos arteriales agudos. La elevada frecuencia de mutaciones en este locus está relacionada a la expresión tardía de los efectos deletéreos de estas mutaciones, después de la etapa reproductiva. Encontramos habitualmente al paciente heterocigota con solo un gene afectado y dominante. Es mucho más infrecuente la presentación homocigota (se estima 1 en 1.000.000), pero la debemos considerar como posibilidad diagnóstica cuando las cifras de colesterol LDL superan los 500 mg/dL en adultos y los 420 mg/dL en niños y jóvenes, especialmente en hijos de parejas consanguíneas (4). Es probablemente más frecuente el heterocigota compuesto, con mutaciones distintas provenientes de dos grupos familiares afectados. La heterogeneidad en la presentación clínica y bioquímica depende de la naturaleza de la mutación, existiendo una correlación importante entre la actividad residual del receptor y la concentración de LDL-colesterol.
Las mutaciones en el gene codificante de la lipoproteína ApoB, la proteína estructural del LDL, se encuentra en una minoría de casos pero llegan a una frecuencia de 1 en 1.000 en descendientes de familias centroeuropeas y celtas. Por su facilidad de determinación por métodos de amplificación-restricción, se incorpora inicialmente al algoritmo diagnóstico. En nuestra población se han encontrado hasta ahora dos sujetos con la mutación Arg3500Gln (la más frecuente en este gene).
El gene PCSK9 regula la degradación intracelular del receptor de LDL y las mutaciones con ganancia de función son capaces de disminuir su densidad en la superficie celular, afectando el clearance plasmático del cLDL y generando un fenotipo semejante a la HF. Las mutaciones con pérdida de función bajan constitutivamente el nivel de cLDL plasmático y el riesgo de enfermedad coronaria por lo que ha generado gran interés la posibilidad de regular farmacológicamente su función.
LOS ESTUDIOS INTERNACIONALES: EL SUBDIAGNóSTICO
En 1974 el grupo de Fredrickson publicó en Circulation resultados de la evaluación de la aparición de enfermedad coronaria (EC) en 116 familias con hiperlipoproteinemia tipo II (según la clásica clasificación de Fredrickson), mostrando la altísima frecuencia de aparición de eventos coronarios asociada a los altos niveles de colesterol. Encontraron que en el varón portador, la probabilidad acumulada de enfermedad coronaria no fatal o fatal (la expectativa de eventos), era de 16% (1 en 6) a la edad de 40 años y pasaba a 52% (1 en 2) a los 60 años. Mientras que en los no portadores ese riesgo era de 12% a los 60 años, menor al riesgo de 20 años atrás de los portadores. En las mujeres portadoras el riesgo se estimó en 32,8% a los 60 años, comparado con un 9,1% a la misma edad en las mujeres normales (5).
Si bien los resultados en esa época fueron muy importantes para establecer la relación entre la hipercolesterolemia y el infarto, que todavía se discutía, no fue hasta dos décadas más tarde que se mostró la incidencia poblacional de la HF y el subdiagnóstico existente. Recién en 1994, Roger R. Williams lanzó el proyecto MEDPED (del inglés: Make Early Diagnosis to Prevent Early Death) en Estados Unidos y sensibilizó a la Organización Mundial de la Salud (OMS) para su lanzamiento mundial, concluyendo que a pesar de los avances en la comprensión de la HF, la mayor parte de las personas no estaban diagnosticadas ni adecuadamente tratadas, lo que resultaría en eventos coronarios tempranos, muchos de ellos fatales. Comenzó una campaña de salud pública y medicina preventiva que se extendió a Europa y otros continentes (6). Los programas de HF de Holanda, España, Reino Unido, Noruega, Austria y Estados Unidos se incorporaron a la International Cholesterol Foundation - InterChol (ICF), que difunde los resultados de más de 40 países. La ICF ya comenzó a trabajar con Australia, Dinamarca, Alemania, Hong Kong, Indonesia, Japón, Corea, Malasia, Nueva Zelandia, Filipinas y Uruguay, en la sensibilización de los gobiernos, el público y las instituciones sanitarias sobre los peligros y los costos de mantener a los pacientes portadores de HF sin diagnóstico ni tratamiento (7).
En el 2000, el grupo de Durrington estableció la utilidad del “hallazgo familiar de casos”, identificando 121 casos nuevos del análisis genealógico testando los familiares de primer grado de 200 pacientes y mostró que hubiera sido necesario el screening poblacional de 60.000 individuos para identificarlos (8). El programa holandés utilizó más tempranamente la identificación extendiéndola a todos los núcleos familiares, al clan o “familia extendida” (9). También quedó clara la inconveniencia de la estimación de riesgo utilizando las ecuaciones de riesgo tipo Framingham, que en estos casos subestiman el riesgo a 10 años.
En el 2001 se publicaron los primeros resultados del programa holandés, revisando cinco años de screening familar (10), que logró la identificación molecular de 2.039 heterocigotas a partir de 237 casos índice, destacando la efectividad del hallazgo de casos basado en la familia (family based case finding) y el diagnóstico molecular. El 50% de los portadores desconocía su hipercolesterolemia, 61% no recibía tratamiento con estatinas y en 18% no se hubiera llegado al diagnóstico con el perfil lipídico. En el reciente congreso de la European Atherosclerosis Society 2010, ya sobre 1.338 casos índice y 11.136 parientes identificados con HF, comunican 85% de pacientes tratados, 80% con estatinas y estiman haber evitado 2.045 eventos de EC (el 71%) de los esperados en esta población, reafirmando la efectividad del programa de prevención (11).
En 2006 se destacó la HF como una causa común y poco reconocida de enfermedad cardiovascular temprana (12), y en el 2008, Humphries y colaboradores (13) discutieron la importancia del diagnóstico en la prevención primaria, mostrando que se podía ganar en promedio nueve años de vida con el tratamiento adecuado y que la mortalidad estandarizada en los pacientes HF entre los 20 y 59 años había bajado de 8,1 a 3,7 después del advenimiento de las estatinas.
Los estudios también mostraron que la mayoría de los pacientes no están advertidos de su condición o no la perciben como riesgosa y permanecen sin tratamiento adecuado a pesar de los regímenes de tratamiento ampliamente conocidos. Si bien el colesterol alto es percibido como un factor de riesgo, los pacientes tienden a enfrentar los altos valores de la HF con medidas dietético-sanitarias propias de dislipemias menos severas, evitando la medicación específica que le salva años de vida. La evaluación de la actuación médica también mostró que las especialidades no estaban jerarquizando el riesgo de estos pacientes, lo que generaba un inadecuado seguimiento y una pérdida de años/medicación. En Europa se estimó que solo 20% de los pacientes portadores estaban diagnosticados y tanto los profesionales como los sistemas sanitarios requerían adecuar la percepción de las dislipemias genéticas como un sector de riesgo y prevención.
El programa español, por su lado, organizó en 2010 un Foro Internacional de Política Sanitaria bajo el lema Hipercolesterolemia familiar: un modelo de atención sanitaria, que mostró el impacto del programa en la identificación de los 100.000 españoles que se estima son portadores de HF. Paradójicamente, la Estrategia Nacional de Prevención Cardiovascular, no incluye a la HF como factor de riesgo. España desarrolló el primer “chip” de ADN para el diagnóstico de HF y copreside la ICF. El Dr. Pedro Mata expuso los resultados de este programa en el pasado XXIV Congreso Sudamericano de Cardiología (2010), realizado en Montevideo (14).
LA SITUACIóN EN URUGUAY: UN ANáLISIS PRIMARIO EN NUESTRA POBLACIóN
La situación en nuestro país resultó muy similar a la resumida en las conclusiones de la OMS. Después de una primera evaluación en 31 afectados provenientes de cuatro familias no emparentadas, solo ocho pacientes con eventos o intervenciones coronarias previas estaban en un tratamiento hipolipemiante aunque inadecuado. El resto de los portadores no tenía diagnóstico de su enfermedad, ni tratamiento farmacológico establecido aún con valores de colesterol total superiores a 300 mg/dl (15). El diagnóstico, en todo caso, alcanzaba a los pacientes en el primer evento coronario entre los 40 y los 50 años y solo los cuatro pacientes índice y algunos hermanos estaban al tanto del diagnóstico y habían adoptado conductas preventivas (16).
Desde al año 2003, la CHSCV trabajó en un programa piloto de evaluación de un modelo de registro nacional de HF, centralizado en el análisis genealógico, el desarrollo del diagnóstico molecular y el asesoramiento genético de estos pacientes en interacción con los equipos médicos tratantes. Los resultados se obtuvieron de 71 casos índice que generaron 71 genealogías médicas con un promedio de tres núcleos familiares, analizando entre tres y cinco generaciones (tabla 2 y figura 1). La población total relevada fue de 1.024 individuos que incluyeron 264 fallecidos y 760 vivos. La población accesible, definida por familiares vivos rastreables eventualmente accesibles al registro fue de 423. El total de la población en riesgo presumible por parentesco y posibilidades de transmisión mendeliana fue de 335 individuos. De esta población se identificaron 294 afectados, de los que 245 están vivos. La calificación de afectado corresponde a los individuos referidos por memoria familiar con datos de historia clínica, colesterol elevado, intervenciones coronarias, muerte cardiovascular prematura o muerte súbita. Esto generó un promedio de 14,4 individuos relevados por caso índice con 4,1 afectados por caso índice, números esperados del método en cascada o seguimiento familiar. Para mejor definición, el método generó 245 afectados vivos, lo que posibilita un hallazgo de 3,4 afectados adicionales por cada caso índice que recibimos. De los 94 niños menores de 15 años relevados en total, 35 se identificaron hasta ahora con probable HF (37%), lo que generó la necesidad de establecer nuevos criterios de edad de inicio del tratamiento, dosis y tipo de intervención, farmacológica y dietética en esta nueva población infantil.

Figura 1. Genealogía médica de una familia portadora de HF.
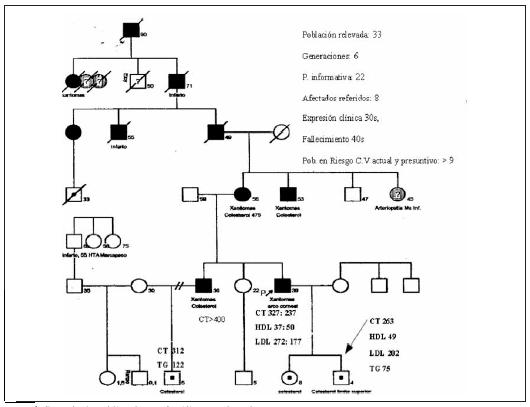
La experiencia del programa piloto permitió establecer la positiva recepción de parte de los pacientes y sus grupos familiares con una clara comprensión del riesgo cardiovascular, con un franco mayor apego a la medicación y un cambio conductual muy definido hacia las conductas sanas preventivas especialmente en el varón joven. El seguimiento de estos pacientes nos permitirá establecer mejor estos aspectos y el establecimiento de nuevos roles familiares en el cuidado del portador HF. El cambio positivo de conductas es uno de los principales argumentos del diagnóstico molecular, seguramente basado en la percepción de la individualidad y la particularidad familiar así como la percepción de la curabilidad de la enfermedad cardiovascular (17).
DIAGNóSTICO DE LAS DISLIPEMIAS AUTOSóMICAS DOMINANTES
Los criterios clínicos para identificar los pacientes con HF están basados en: historia familiar de hipercolesterolemia (en especial en niños), concentraciones altas de cLDL, depósitos en tejidos extravasculares como xantomas tendinosos, xantelasmas periorbitales y arco corneal, y fundamentalmente una historia personal y familiar de enfermedad coronaria prematura.
Los pacientes heterocigotos tienen habitualmente concentraciones de cLDL que duplican los valores normales, en un rango de cLDL desde 190 a 500 mg/dl. Los triglicéridos plasmáticos se encuentran en el rango de normalidad y esto es aprovechado por los criterios de inclusión para diferenciar la HF de la hipercolesterolemia combinada familiar y otras dislipemias más frecuentes no dominantes (fundamentalmente multifactoriales). Sin embargo, algunos pacientes pueden tener los triglicéridos elevados como consecuencia de interacción con otros genes (genotipo ApoE: E2/E2) o con factores ambientales (alcohol, sobrepeso, sedentarismo y diabetes mellitus [DM2]).
Entre los signos físicos los xantomas tendinosos en tendón de Aquiles y extensores de la mano son patognomónicos de la HF, pero solo están presentes en menos de 30% de los pacientes. Son un fuerte indicador cuando están presentes antes de los 45 años (tabla 3).

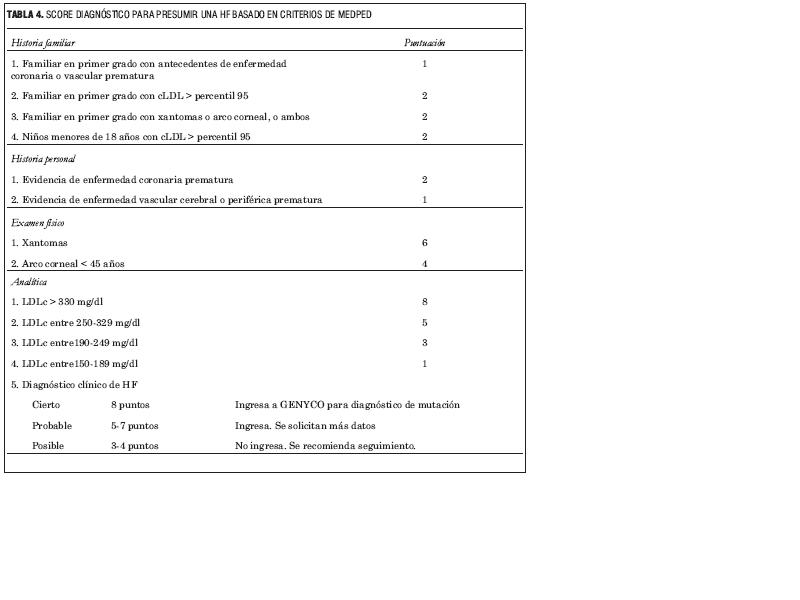
Como no existen criterios clínicos predictivos absolutos para la HF y existe, sin embargo, un número tan significativo de portadores, se han ensayado sistemas de score que buscan una identificación rápida. El sistema de puntuación MEDPED está basado en la experiencia holandesa y es recomendado en Europa y utilizado por el programa HF español, y ha sido adoptado en GENYCO para su evaluación. Este criterio asigna puntaje teniendo en cuenta la historia familiar de cLDL, de enfermedad cardiovascular, la presencia de arco corneal antes de los 45 años y los xantomas y categoriza al paciente en cuatro grupos de concentración de cLDL (desde 150 a más de 300 mg/dl) sin hipertrigliceridemia. Esta puntuación (tabla 4) tiene la ventaja de ser fácil de utilizar en la práctica diaria pero tiene el inconveniente de que no efectúa un diagnóstico inequívoco. La falta de datos o los datos incompletos basados en la memoria familiar pueden colocar a los pacientes en las categorías de “posible” o “probable”. Se debe destacar que el programa español identificó como portadores de mutación 40% de los pacientes en estas categorías, mostrando las limitaciones eventuales del score MEDPED (18). El programa GENYCO está evaluando el score MEDPED y otros formatos de recolección rápida de datos diagnósticos que se adapten mejor a la clínica práctica. En este caso el paciente y su grupo familiar pueden no acceder a GENYCO o al diagnóstico molecular que es el eje del diagnóstico positivo. En estos pacientes es importante el seguimiento clínico y la revisión periódica del caso. El cardiólogo debe utilizar su propio criterio clínico si está frente a la convicción de un caso cierto con insuficiente información.
EL DIAGNóSTICO MOLECULAR
El desarrollo rápido de la capacidad del diagnóstico molecular lo estableció como el gold standard de esta patología. La proliferación y el abaratamiento de los costos de secuenciación de ADN mejoraron las perspectivas de determinación directa de las mutaciones en el paciente y así la etiopatogenia de la dislipemia. Además la demostración que 23,5% con mutación está por debajo del percentil 90 de LDL y 14,7% sin mutación está por encima del percentil 90, reveló que la consideración de la concentración de LDL era un pobre predictor de mutación en el RLDL (19,20).
El diagnóstico basado en el análisis del gene RLDL genera resultados negativo/positivo, altamente específicos, recomendados por la OMS en el programa MEDPED.
En poblaciones como la nuestra, donde existe un gran número de mutaciones causantes de HF distribuidas a lo largo de los 18 exones del gene, el promotor y las regiones intrón-exón, el diagnóstico genético resulta laborioso en el primer caso seleccionado de cada grupo familiar cuando la mutación es desconocida. Una vez encontrada, el diagnóstico se simplifica a la amplificación del exón involucrado para todos los miembros de la familia.
El algoritmo seguido para el diagnóstico molecular incluye: 1) el testado de mutaciones en ApoB y ApoE, que descartan la HF por ApoB defectuosa y la hiperlipoproteinemia tipo III por homocigosis de la variante E2 de ApoE. Además informa sobre la presencia de la variante ApoE4, que es un factor de riesgo independiente de EC y modificador eventual de la velocidad de progresión de la aterosclerosis, así como un factor a considerar para un comienzo más temprano del tratamiento hipolipemiante; 2) el testado de mutaciones del gene RLDL (21).
El procedimiento general implica la amplificación de ADN por el método de PCR (reacción en cadena de la polimerasa) a partir de una muestra de sangre periférica y la secuenciación de los fragmentos específicos seguida de la verificación de mutaciones contra las secuencias de referencias contenidas en bases de datos específicas.
EL DIAGNóSTICO DIFERENCIAL CON OTRAS DISLIPEMIAS FAMILIARES NO DOMINANTES
Debemos considerar que el diagnóstico basado en los datos del perfil lipídico, hallazgos físicos y antecedentes familiares de hipercolesterolemia y EC prematura, pueden ser expresión de otras dislipemias aterogénicas familiares entre las que se destacan: la hipercolesterolemia familiar combinada (HFC), la hipertrigliceridemia familiar (HTGF) y las dislipemias mixtas asociadas a la DM2 fundamentalmente y la Lp(a) alta familiar. Esta última, con cifras mayores de 30 mg/dl, la hemos encontrado con una frecuencia relativamente alta en pacientes varones jóvenes con IAM o enfermedad coronaria prematura y cifras en rango del perfil lipídico clásico.
Frecuentemente, existe un solapamiento entre el fenotipo lipídico de la HF y el de la HFC. En estos casos, el diagnóstico diferencial puede requerir el estudio sistemático de varios perfiles espaciados en el tiempo del paciente propósito y, paralelamente, el estudio de los perfiles de familiares de diferente edad y condición física para establecer un diagnóstico presuntivo que nos permita decidir el diagnóstico molecular. La identificación de mutaciones en el receptor de LDL o ApoB definitivamente proveen un diagnóstico inequívoco de HF (22). En estas circunstancias es útil y recomendable el diagnóstico de otros genes asociados a la HFC como las variantes en las apolipoproteínas ApoE, y ApoA5, ambas vinculadas a la susceptibilidad a la hipertrigliceridemia en la HFC y la HTGF, especialmente cuando el desencadenante es el síndrome metabólico y la DM2. La variante A5 S19W de ApoA5 parece que es un franco factor de susceptibilidad genética en el desarrollo de la hipertrigliceridemia cuando actúa sinérgicamente con otras variantes genéticas como la E2 o E4 de ApoE, que modulan el funcionamiento adecuado de las apolipoproteínas en las partículas de lípidos plasmáticos.
PAPEL DE OTROS FACTORES EN LA VALORACIóN DEL RIESGO EN EL PACIENTE JOVEN
El papel de los factores de riesgo establecidos y los factores emergentes puede ser importante en la expresión de la enfermedad. Debemos recordar que en Uruguay el 50% de la población comprendida entre los 25 y 65 años tiene tres o más factores de riesgo. El tabaquismo es el factor principal reconocido, duplicando el riesgo de aparición de la EC. La prevención de HF incorpora tanto la modificación de factores de riesgo conocidos como la identificación de la susceptibilidad genética a la enfermedad. Pero debemos invertir el razonamiento habitual: las consecuencias de la HF no las define el ambiente sino primariamente la genética y, por tanto, las medidas de modificación de los comportamientos de riesgo y medidas dietéticas no alcanzan para la prevención primaria ni secundaria de eventos cardiovasculares. El médico debe educar sobre la necesidad del control farmacológico crónico, permanente de la hipercolesterolemia, frecuentemente con dosis altas, y la combinación eventual de fármacos que sean necesarios para alcanzar los objetivos de cLDL.
El registro genera un alto número de pacientes jóvenes y niños, sin expresión vascular de la enfermedad, en etapa preclínica, por lo que las técnicas menos invasivas de valoración de la aterosclerosis subclínica y otros marcadores biológicos de los llamados emergentes cobran relevancia. La estimación del espesor de íntima-media carotídeo y la búsqueda de placa carotídea, así como la estimación de la disfunción endotelial, forman parte de los exámenes indicados en el paciente joven que va a estar sujeto a una valoración sistemática de la respuesta a los hipolipemiantes.
Los exámenes de homocisteinemia, fibrinogenemia, Lp(a) y PCR deben siempre descartar alteraciones concomitantes de estos valores en pacientes con HF, ya que son factores de riesgo independientes que pueden acelerar la enfermedad aterogénica. El terreno protrombótico debe ser evaluado con sus descriptores hematológicos clásicos y su genética asociada, como las variantes de factor II y factor V, MTHFR y PAI-1. Entre las afecciones más agravantes están las que encontramos hoy como epidémicas y muy dependientes de estilos de vida como la hipertensión arterial, la diabetes mellitus, el bajo cHDL.
TRATAMIENTO
El tratamiento de los pacientes con HF depende de factores comunes, como la edad, y de factores de riesgo, comorbilidades y fundamentalmente del estado evolutivo de la aterosclerosis, especialmente coronaria. Con estas consideraciones el tratamiento progresa generalmente desde las medidas higiénico- dietéticas en la infancia al tratamiento farmacológico precoz con estatinas o tratamiento combinado con otros hipolipemiantes en la edad adulta, seguido eventualmente de cirugía vascular coronaria. La referencia a especialistas en el tema es importante en el establecimiento de las dosis efectivas y seguras de hipolipemiantes.
La mayoría de los pacientes con HF no alcanza los objetivos de cLDL <100 mg/dl, con medidas exclusivamente higiénico-dietéticas, que solo bajan los niveles de cLDL entre 10% y 15% cuando se requieren respuestas de 50% o más. La administración de fármacos hipolipemiantes es la regla y frecuentemente en forma agresiva con indicaciones que pueden cuadruplicar las dosis estándar de estatinas. Como destacamos antes, la identificación de niños genera la necesidad de nuevos criterios de edad de inicio del tratamiento, dosis y tipo de intervención farmacológica y dietética en esta nueva población infantil. Varias guías específicas, como las de NICE (23) o la del registro inglés de HF Simon Broome (24), han propuesto criterios de tratamiento vinculados a la estimación del riesgo individual y serán objeto de una comunicación específica.
EL PROGRAMA GENYCO
GENYCO intenta resumir la experiencia internacional y nacional en un programa de detección de población con HF mediante el diagnóstico genético y una búsqueda familiar activa. El programa sigue las recomendaciones de la OMS, utilizando el modelo europeo de identificación de pacientes y los lineamientos de la Comisión Honoraria para la Salud Cardiovascular en el sentido de promover, coordinar y desarrollar planes y programas concernientes a la prevención, diagnóstico precoz, tratamiento y rehabilitación de personas expuestas o afectadas por enfermedades cardiovasculares.
El programa intenta revertir la situación de subdiagnóstico y subtratamiento de una enfermedad crónica, sensible a la acumulación de factores de riesgo ambiental, biológico y genético, que actúan en forma acumulativa con el correr de los años. En Uruguay, el 50% de la población comprendida entre los 25 y 65 años tiene tres o más factores de riesgo, por lo que tiene riesgo elevado de sufrir EC y morir por esta causa. El tratamiento farmacológico de la hipercolesterolemia con estatinas y otros hipolipemiantes, basado en el conocimiento de las bases moleculares de esta enfermedad, produce una reducción sustancial del riesgo de EC prematura y puede lograr una sobrevida similar al resto de la población no afectada.
La HF es actualmente el paradigma de la medicina molecular y preventiva. En los hechos se ha promocionado internacionalmente como un excelente modelo para demostrar los beneficios de la prevención de la enfermedad coronaria y el infarto precoz en el adulto joven, identificando fácilmente un grupo poblacional con un elevado riesgo genético y utilizando la caracterización molecular de mutaciones extendidas en nuestras poblaciones. Los cardiólogos en particular deben reconocer a estos pacientes y grupos familiares y utilizar la oportunidad de una intervención más temprana y más individualizada. La tragedia de la muerte del adulto joven por enfermedad coronaria es razonablemente prevenible y evitable.
BIBLIOGRAFíA
1. Marks D, Thorogood M, Neil HA, Humphries SE. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis 2003;168:1-14.
2. Goldstein JL, Brown MS. The LDL receptor defect in familial hypercholesterolemia. Implications for pathogenesis and therapy. Med Clin North Am 1982; 66: 335-62.
3. Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolaemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular basis of inherited disorders. 7th ed. New York: McGraw Hill, 1995: 19812030.
4. Esperón P, Raggio V, Stoll M. Una nueva mutación en el promotor del gene del receptor de LDL asociada con hipercolesterolemia familiar en homo y heterocigosis. Clínica e Investigación en Arteriosclerosis 2009; 21: 51-5.
5. Stone NJ, Levy RI, Fredrickson DS, Verter J. Coronary artery disease in 116 kindred with familial type II hyperlipoproteinemia. Circulation 1974; 49: 476-88.
6. WHO. Human Genetics Program. Familial hypercholesterolaemia, a global perspective. Ginebra: WHO, 1999.
7. International Cholesterol Foundation. Communiqué and Press Release. [comunicado de prensa en Internet]. The International Cholesterol Foundation (ICF). Obtenido de: http://www.interchol.org/post/Communique-and-Press-Release-The-International-Cholesterol-Foundation-ICF/ Consultado (18/5/2011).
8. Bhatnagar D, Morgan J, Siddiq S, Mackness MI, Miller JP, Durrington PN. Outcome of case finding among relatives of patients with known heterozygous familial hypercholesterolaemia. BMJ 2000; 321: 1497500.
9. Kastelein JP. Screening for familial hypercholesterolaemia Effective, safe treatments and DNA testing make screening attractive. BMJ 2000; 321: 1483–4.
10. Umans-Eckenhausen MA, Defesche JC, Sijbrands EJ, Scheerder RL, Kastelein JJ. Review of first 5 years of screening for familial hypercholesterolaemia in the Netherlands. Lancet 2001; 357: 165-8.
11. Huijgen R, Versmissen J, Oosterveer DM. Efficacy of 15 years of genetic cascade screening for familial hypercholesterolemia in the Netherlands in prevention of coronary artery disease [abstract]. European Atherosclerosis Society 2010; 11(Suppl.2): S12.
12. Yuan G, Wang J, Hegele RA. Heterozygous familial Hypercholesterolemia: an underrecognized cause of early cardiovascular disease. Can Med Assoc J 2006; 174: 1124-9.
13. Humphries SE, Hadfield G. Identifying patients with familial hypercholesterolaemia in primary care. Heart 2008; 94: 695-6.
14. Mata P. Hipercolesterolemia familiar: un modelo para la prevención de la enfermedad cardiovascular. La experiencia española. Congreso Sudamericano de Cardiología, 24. Montevideo, 2010.
15. Stoll M, Raggio V. La historia familiar como instrumento de prevención en la enfermedad cardiovascular. Boletín de la Comisión Honoraria para la Salud Cardiovascular 2002; 4: 43-9.
16. Stoll M, Esperón P, Raggio V. Progresos en la identificación de pacientes con Hipercolesterolemia Familiar (HF). Avances hacia un registro nacional de HF. Boletín de la Comisión Honoraria para la Salud Cardiovascular 2005; 6: 42-3.
17. Vergopoulos A, Knoblauch H, Schuster H. DNA testing for familial hypercholesterolemia: improving disease recognition and patient care. Am J Pharmacogenomics 2002; 2: 253-62.
18. Pocoví Mierasa MD, Tejedor Hernández B. Diagnóstico genético de la hipercolesterolemia familiar. JANO 2005; 69: 41-4.
19. Damgaard D, Larsen ML, Nissen PH, Jensen JM, Jensen HK, Soerensen VR, et al. The relationship of molecular genetic to clinical diagnosis of familial hypercholesterolemia in a Danish population. Atherosclerosis 2005; 180: 155-60.
20. Civeira F, Ros E, Jarauta E, Plana N, Zambon D, Puzo J, et al. Comparison of genetic versus clinical diagnosis in familial hypercholesterolemia. Am J Cardiol 2008; 102: 1187-93-1193.e1
21. Williams RR, Hunt SC, Schumacher MC, Hegele RA, Leppert MF, Ludwig EH, Hopkins PN. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am J Cardiol 1993; 72: 171-6.
22. Carmena R, Roy M, Roederer G, Minnich A, Davignon J. Coexisting dysbetalipoproteinemia and familial hypercholesterolemia. Clinical and laboratory observations. Atherosclerosis 2000; 148: 113-24
23. Chowdhury TA, Hitman GA. NICE guidance for identification and treatment of familial hypercholesterolaemia: Commentary 1 Heart, 2009; 95: 587-9.
24. Scientific Steering Committee on behalf of the Simon Broome Register Group. Mortality in treated heterozygous familial hypercholesterolaemia: implications for clinical management. Atherosclerosis 1999; 142: 105-12.


{kind=link}