










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkRevista Uruguaya de Cardiología
versión On-line ISSN 1688-0420
Rev.Urug.Cardiol. vol.22 no.1 Montevideo jun. 2007
Estratificación del riesgo en la miocardiopatía hipertrófica
DRES. GUILLERMO ROMERO-FARINA (Ph.D, FESC, FASNC), JAUME CANDELL-RIERA (Ph.D, FESC)
Servicio de Cardiología. Hospital Universitari Vall d'Hebron. Barcelona. España.
Correspondencia: Jaume Candell-Riera, Guillermo Romero-Farina. Servicio de Cardiología, Hospital Universitari Vall d'Hebron, Universitat Autònoma de Barcelona. Paseo Vall d'Hebron 119-129, 08035 Barcelona, España. Jcandell@vhebron.net; guirom@telefonica.net
Abreviaturas: FA: fibrilación auricular; FE: fracción de eyección; IC: insuficiencia cardíaca; MH: miocardiopatía hipertrófica; MS: muerte súbita; TV: taquicardia ventricular; TVNS: taquicardia ventricular no sostenida; TVS: taquicardia ventricular sostenida; VFC: variabilidad de la frecuencia cardíaca.
Recibido abril 18, 2007; aceptado abril 27, 2007.
PALABRAS CLAVE: CARDIOMIOPATÍA HIPETRÓFICA
TOMOGRAFÍA DE EMISIÓN
COMPUTARIZADA DE FOTÓN SIMPLE
REVISIÓN [TIPO DE PUBLICACIÓN]
KEY WORDS: CARDIOMIOPATHY,
HYPERTROPHIC
TOMOGRAPHY,
EMISIÓN-COMPUTED, SINGLE-PHOTON
REVIEW [PUBLICATION TYPE]
En la década de 1960 se describió la miocardiopatía hipertrófica (MH) como la causa más común de muerte súbita (MS) cardíaca en gente joven y atletas competitivos (1-3). Ahora bien, muchos pacientes sintomáticos pueden permanecer clínicamente estables por años y otros pueden desarrollar una historia natural sin que aparezcan síntomas (4-6).
Un factor que dificulta el análisis de la historia natural de esta enfermedad es que la MH no es una enfermedad frecuente y que la mayor parte de las publicaciones corresponden a series sesgadas y relacionadas con pacientes referidos a centros terciarios de EEUU, Inglaterra, Japón, Canadá, Alemania e Italia (7).
El 50% de 78 estudios de MH durante un período de cinco años (desde 1983 a 1987) fue publicado en Circulation, The American Journal of Cardiology, The Journal of the American College of Cardiology y en British Heart Journal. En treinta y cinco (45%) de estos 78 estudios se habían seleccionado 2.483 (73%) de los 3.404 pacientes totales con MH, y estos provenían solamente de dos centros: el National Heart, Lung and Blood Institute (Bethesda) y el Royal Postgraduate Medical School (Hammersmith, London) (7,8).
La mayor parte de los pacientes con MH son referidos a los centros terciarios debido a que presentan síntomas, existiendo, por tanto, un sesgo importante en las poblaciones estudiadas (1,9-11). Estas consideraciones deben tenerse en cuenta cuando los resultados de estos trabajos se pretenden aplicar a pacientes asintomáticos o poco sintomáticos (1,12-15).
La muerte prematura inesperada es una de las posibilidades de la historia natural de estos enfermos (1,3,16-24). Sin embargo, no todos los pacientes con MH tienen el mismo riesgo de muerte prematura (6,23-25) y, por tanto, existe una investigación intensa dirigida hacia la identificación de factores de riesgo de MS (1,11,23,24,26-31).
La incidencia de MS entre los pacientes con MH varía según las series publicadas (7,30). Así, en series que provienen de centros de referencia, se encuentran tasas de MS entre 3% y 6% por año. Sin embargo, en poblaciones no seleccionadas la tasa publicada de MS no supera 1% por año. La mortalidad cardíaca total registrada en nuestra población, estudiada durante un seguimiento medio de diez años, fue de 5,9%, con una mortalidad anual de 0,6% (0,006 muertes por persona por año), significativamente mayor al compararla con la población general catalana (figura 1) y sin diferencias entre hombres y mujeres (32).
Figura 1. Curvas Kaplan-Meier de sobrevida entre la población catalana (línea de cuadrados negros) y los pacientes con miocardiopatía hipertrófica (línea con rombos negros).
La MS aparece con más frecuencia en niños y adultos jóvenes, entre 14 y 35 años de edad (33), aunque también puede ocurrir en adultos y sería extremadamente rara en niños menores de 5 años (34). Una proporción importante de MS aparece durante o inmediatamente después de un esfuerzo vigoroso (35). Ello ha hecho de la MH la primera causa de muerte en atletas jóvenes, lo que desaconseja totalmente la práctica de deportes de competición en los portadores de la enfermedad. En los estudios con seguimiento medio superior a cuatro años y que incluyen más de 100 pacientes, la mortalidad cardíaca de la MH osciló entre 4,6% y 25% (valor medio 13,6%), con una mortalidad anual entre 0,6% y 3,4% (tabla 1) (28,31,32,36-45), menor a la observada en adolescentes y jóvenes (4% al 5,8% anual) (21,22,28).
Tabla 1
Las dos causas más frecuentes de muerte en esta enfermedad son la MS y la insuficiencia cardíaca (IC), con un promedio de MS anual de 1,8% y de muerte por IC de 0,5%. En la mayoría de estos estudios la muerte súbita es la causa más frecuente de fallecimientos, correspondiendo a más del 50% de las muertes de origen cardíaco (entre 55% y 86%), y a más del 30% de las muertes totales (entre 35% y 75%) sobre todo en niños y jóvenes. La muerte por IC ocurre entre 3% y 50%, con excepción de la serie de Cecchi y colaboradores (39), donde corresponde a 78% de las muertes y la MS a 14%.
Estos resultados se asemejan a nuestra serie, donde la causa más frecuente de muerte fue la IC (57%). El 71% (cinco pacientes) de las muertes en nuestra población ocurrieron antes de los 75 años. Elliott y colaboradores (46) consideran muerte prematura la que ocurre antes de los 40 años.
Maron y colaboradores (45) analizaron una extensa serie de 744 pacientes durante un seguimiento medio de 8,7 años y hallaron una mortalidad de 12%; 51% de éstas correspondían a MS, 36% a IC progresiva y 13% a accidentes cerebrovasculares. La MS se presentaba en pacientes más jóvenes (45 +/- 12 años), mientras que la ocasionada por IC y accidente cerebrovascular se presentaba más tardíamente (56+/-19 y 73+/-14 años, respectivamente).
ESTRATIFICACIóN DE RIESGO
Uno de los principales objetivos a evaluar en los pacientes con MH es la identificación del riesgo individual de MS (47). En los pacientes jóvenes el síncope, la historia familiar de MH y de MS prematura son predictores específicos pero poco sensibles de MS (5,6). En pacientes mayores la presencia de taquicardia ventricular (TV) no sostenida en el Holter agrega información para estratificar el riesgo, pero es un débil predictor de muerte cardíaca (7). Además de los datos clínicos existen diferentes técnicas utilizadas para la estratificación del riesgo en la MH, aunque no todas han demostrado ser eficaces.
Del análisis de los datos clínicos y de parámetros derivados de estas técnicas se han extraído diferentes variables clínicas y de laboratorio para la estratificación de riesgo. Maron y colaboradores (30) las agruparon en factores de riesgo probables y potenciales asociados a MS en la MH, pero en la actualidad las guías terapéuticas norteamericanas y europeas definen claramente los factores de riesgo mayores y posibles (48) (tabla 2) de MS debido al beneficio pronóstico que tienen los cardioversores-desfibriladores implantables.

Se han realizado numerosos trabajos clínicos con diferentes diseños, prospectivos (39,40,42,49,50), retrospectivos (28,31,41,44,51), de población intrahospitalaria (40,42,52-54) y extrahospitalaria (8,31,39), en pacientes asintomáticos (42,39,55) y sintomáticos (55), con (49,56,38) y sin obstrucción dinámica (49,57) con el objetivo de evaluar las variables pronósticas durante la historia natural de la enfermedad (28,39,40,42,56-58).
En las tablas 3 (9,28,31,39,40,49,54) y 4 (32,41-44,59,60) se resumen las publicaciones más recientes que incluyen como mínimo 100 pacientes y con un período de seguimiento superior a los cinco años. Las variables pronósticas derivadas de estos estudios se exponen en la tabla 5 (1,2,24,28,29,32,33,38-44,49,50-54,59-68).
Tabla 4
VARIABLES DERIVADAS DE LA CLíNICA
La presencia de historia familiar de MH y MS, sobre todo en jóvenes, es más frecuente en los pacientes que han fallecido (69,70). Actualmente está demostrado que el antecedente de historia familiar maligna de MH es de mal pronóstico, y se define historia familiar maligna a la presencia de MS en dos o más familiares de primera generación (11,71).
La edad en el momento del diagnóstico es una variable predictora de peor pronóstico. La supervivencia es menor en aquellos pacientes en los que el diagnóstico se ha realizado cuando su edad era inferior a 14 años (69).
El antecedente de ser sobreviviente a una MS es otra variable predictora independiente de nuevos eventos cardíacos y decisiva para la indicación de un cardiodesfibrilador (72-74).
En varios estudios la disnea clase funcional III-IV en el momento del diagnóstico es otra variable predictora de mal pronóstico (75). En nuestros resultados la disnea severa (clase funcional III-IV) no fue predictora de mortalidad, pero sí lo fue para la colocación de marcapaso cardíaco al igual que la edad superior a 50 años. También hallamos que la angina de grado severo era predictora de miotomía-miectomía quirúrgica.
En niños y adolescentes el presíncope y el síncope recurrente parecen identificar a enfermos con mayor riesgo de MS. Sin embargo, en contraste con la estenosis aórtica valvular, estos síntomas pueden no ser un signo ominoso en adultos; algunos pacientes tienen antecedentes de estos episodios muchos años atrás, sin que se hayan repetido posteriormente (76).
VARIABLES DERIVADAS DE TéCNICAS ELECTROCARDIOGRáFICAS
Electrocardiograma de superficie
El electrocardiograma de superficie no parece una técnica adecuada para estratificar el riesgo de MS en la MH, pero se ha observado que en los pacientes asintomáticos con evidencia electrocardiográfica de hipertrofia ventricular izquierda, la presencia de arritmia ventricular frecuente o compleja se asocia con un incremento de la mortalidad (43). El pronóstico en relación con la mortalidad y la severidad de los síntomas generalmente no difiere entre los pacientes con o sin inversión de la onda T (77), mientras que los cambios ST-T y las extrasístoles ventriculares se relacionan con riesgo de fibrilación auricular (FA) (78).
Otro hallazgo electrocardiográfico con impacto pronóstico es la presencia de FA. La FA se observa entre 10% y 28% de los pacientes con MH (79,25) y se ha asociado con deterioro de la función ventricular, edema pulmonar, angina, síncope y complicaciones embólicas (17,80-82). Datos de diferentes centros (28,79,83,84) sugieren que aproximadamente 5% de los pacientes tienen FA en el momento del diagnóstico y que después de cinco años de seguimiento se observa en 10% adicional de pacientes. Parece existir una relación estrecha entre la velocidad del flujo en la orejuela izquierda y la función contráctil de la aurícula izquierda con riesgo elevado de accidente cerebrovascular embólico en pacientes con MH y FA paroxística (85). Stafford y colaboradores (86) reportaron un paciente de 15 años (antecedentes de paro cardíaco por fibrilación ventricular) que durante el estudio electrofisiológico se inducía FA con respuesta ventricular rápida asociada a hipotensión y evidencia de isquemia miocárdica, que acababa en fibrilación ventricular.
En otro estudio (87) se observó que la clase funcional se deteriora en 74% de los pacientes después de su primer episodio de FA. El 63% de los pacientes retornan a ritmo sinusal con tratamiento farmacológico y 24% con cardioversión eléctrica adicional. Estos autores defienden que la FA no representa un signo ominoso en la historia natural de la MH. No obstante, estas conclusiones son contradictorias con otros estudios (88), donde se ha notado una mayor mortalidad con el desarrollo de FA. Esto último concuerda con nuestros resultados, pues en el análisis estadístico bivariable la presencia de arritmia completa por FA en el electrocardiograma fue una variable pronóstica de mortalidad (32).
Más recientemente, en un estudio epidemiológico japonés (59) de 2.155 pacientes, con un seguimiento de cinco años en 1.605 de ellos, se observó que el bloqueo de rama izquierda del haz de His en el análisis multivariante constituye una de las variables predictoras independientes de mortalidad cardíaca, datos no hallados en la población occidental.
Promediación de señales
Es un método que evalúa los potenciales de la porción final del complejo QRS y del segmento ST (41). Los potenciales tardíos se asocian con una predisposición a la TV y MS en diferentes enfermedades cardíacas, incluyendo la cardiopatía isquémica después de un infarto de miocardio y la miocardiopatía dilatada (89-92). Kulakowski y colaboradores (93) evaluaron el valor de la denominada 'signal-averaged electrocardiography' (SAEG) para identificar pacientes con MH de alto riesgo de MS y concluyeron que no era útil para identificar pacientes con arritmia ventricular o riesgo elevado de MS. La incapacidad del SAEG para estratificar el riesgo probablemente se deba a diferentes factores:
1. El sustrato arritmogénico miocárdico en la MH es un proceso fibrótico más difuso, a diferencia de la cicatriz del infarto agudo de miocardio que es más localizada, y representa el sustrato anatómico de la TV (47,94).
2. La hipertrofia ventricular de la MH puede enmascarar los potenciales tardíos (47,94).
3. Los potenciales tardíos son un marcador de TV monomorfa sostenida, la cual es rara en la MH (47).
4. La MS también está provocada por otras causas: isquemia, bradicardia, taquicardia supraventricular y obstrucción severa en el tracto de salida del ventrículo izquierdo (12,21,28,95-97).
Dispersión y prolongación del intervalo QT
El intervalo QT representa la repolarización o la recuperación del miocardio excitado y la dispersión QT es la variación del intervalo QT en el electrocardiograma de superficie (98-100). La prolongación del intervalo QT y el aumento de la dispersión QT se han descripto en pacientes con síndrome de QT largo congénito (100-103), en pacientes que reciben agentes antiarrítmicos clase IA, clase III, sotalol (104), amiodarona (101,105), y en pacientes con infarto agudo de miocardio (100,104,106) propensos a desarrollar arritmias ventriculares (98-101).
En la MH la presencia de inestabilidad eléctrica es un determinante importante de MS (19,53,25). El sustrato patológico de esta enfermedad puede ser una condición que provoque prolongación del intervalo QT y una excitabilidad y recuperación no homogéneas (107). En un estudio (108) se encontró que existía una prolongación del intervalo QT y un incremento de la dispersión QTc en los 24 pacientes con MH estudiados comparados con los sujetos normales. Buja y colaboradores (109), estudiando 26 pacientes, encontraron un aumento significativo de la dispersión del QT en pacientes con MH, especialmente en aquellos que tenían arritmias ventriculares. Estos hallazgos contrastan con los de Martin y colaboradores (110). Estos autores, al estudiar retrospectivamente una pequeña población de 25 niños con MH, hallaron que el estudio de dispersión y prolongación del intervalo QT tiene un valor limitado.
Actualmente se intenta relacionar la variabilidad del intervalo QT con la variabilidad de la frecuencia cardíaca, para obtener el índice de variabilidad QT (111,112). Yi y colaboradores (113) estudiaron los intervalos y las dispersiones QT y JT en 46 pacientes con MH (23 con MS y 23 sin MS) y hallaron un incremento significativo en la dispersión de ambos intervalos en los pacientes con MS, al igual que en pacientes con miocardiopatía dilatada cuando son comparados con sujetos normales (114).
Disfunción autonómica
El ritmo cardíaco está bajo la influencia del sistema nervioso autonómico (115). La disfunción autonómica cardíaca se evidencia a través de la disminución de la variabilidad de la frecuencia cardíaca y se asocia con un incremento de la incidencia de arritmias ventriculares y MS en la enfermedad arterial coronaria, especialmente después de un infarto agudo de miocardio (116). También se ha encontrado que la variación de la actividad barorreceptora es expresión de la disfunción autonómica y predictora de arritmias ventriculares y MS.
El análisis de la variabilidad de la frecuencia cardíaca (VFC) ha sido ampliamente aceptado como un método incruento para valorar la influencia del sistema nervioso autónomo sobre el corazón (8). Se ha observado (117-120) una disminución significativa de la VFC en los pacientes con MH con respecto a los sujetos normales. También se ha descrito un descenso en el tono simpático durante las actividades diarias (121,122). Algunos autores (123) señalaron que la alteración del tono autonómico está relacionada con la severidad de la hipertrofia ventricular. Otros autores opinan que el reflejo barorreceptor contribuye a las fluctuaciones de la frecuencia cardíaca y que la VFC alterada podría ser en parte debida a una función sistólica supernormal (124), a una presión aumentada de la aurícula izquierda (reflejo de Bainbridge) y a una activación de los mecanorreceptores en los ventrículos (reflejo de Bezold-Jarisch). La isquemia miocárdica en la MH (125) y el fallo de los reflejos neurovegetativos en el corazón, como consecuencia de la hipertrofia (126), pueden contribuir a los cambios autonómicos en esos pacientes. La VFC podría ser predictora de la IC, aunque no queda claro que lo sea para la MS (127).
Así pues, el análisis de la VFC en pacientes con MH puede ser una herramienta útil para conocer el estado del sistema nervioso autónomo y determinar el grupo con mayor tono simpático, que parece asociarse a mayor frecuencia de arritmias ventriculares y, por lo consiguiente, a un subgrupo de mayor riesgo.
Estimulación ventricular programada
El papel de la estimulación eléctrica programada en la MH ha sido tema de controversia. La estimulación ventricular programada fue utilizada inicialmente para la inducción de TV monomorfa sostenida en la cardiopatía isquémica (128-130), y más adelante diferentes estudios evaluaron el valor de la estimulación ventricular programada en la MH (131-136).
Fananapazir y colaboradores (95) evaluaron los resultados del estudio electrofisiológico en 155 pacientes con MH. La taquicardia ventricular no sostenida (TVNS) fue inducida en 22 pacientes (14%), la taquicardia ventricular sostenida (TVS) en 66 (43%) de las cuales 16 fueron monomorfas, 48 polimorfas y dos fibrilaciones ventriculares. La inducción de una TVS (en la mayoría de los pacientes estudiados) es un hallazgo anormal en la MH que podría servir de guía para la terapéutica, pero la especificidad de esta técnica es muy pobre, lo que hace que sea un método de evaluación inapropiado para el seguimiento y particularmente para decidir el implante de un cardiodesfibrilador automático. La experiencia europea está de acuerdo en que la incidencia de arritmias ventriculares inducidas está en relación con la agresividad del protocolo de estimulación.
De los resultados obtenidos hasta la actualidad no puede deducirse que esta técnica deba aconsejarse de forma generalizada como guía terapéutica y para la estratificación del riesgo en los pacientes con MH.
Estudio de la conducción
Una alternativa electrofisiológica para la estratificación del riesgo es la valoración del desarreglo de las fibras miocárdicas reflejadas en el registro electrofisiológico (135). Ello está basado en la hipótesis de que el incremento del desarreglo miocárdico desarrolla vías de conducción anormales (más largas y tortuosas) vinculadas a la presencia de fibrosis miocárdica. Estas vías presentan características electrofisiológicas diferentes, como, por ejemplo, diferentes velocidades de conducción (137). Esta conducción heterogénea es un componente más del sustrato fisiopatológico de reentrada para la TV o FV, o ambas.
Holter
El mecanismo de la MS no es bien conocido, pero es muy probable que en muchos casos sea de origen arrítmico (23,138,139) y las arritmias ventriculares son frecuentes de observar en el Holter (tabla 6) (9,25,53,58,79,134,140-142).
Varios estudios prospectivos han demostrado que las extrasístoles ventriculares y particularmente la TV se asocian con un elevado riesgo de MS cardíaca (143). Dos estudios (25,53) observaron, en un total de 169 pacientes con MH y con un seguimiento de tres años, que la MS cardíaca ocurrió con mayor frecuencia en pacientes con TV en el Holter en comparación con los pacientes sin esta arritmia. Sin embargo, la TV fue también hallada en 21% de los sobrevivientes. Por otra parte, la mortalidad anual en los niños es de 6%, significativamente más elevada que la de los adultos, pero a pesar de ello la TV es poco frecuente en los niños y no parece ser una variable que sea de utilidad para identificar riesgo de MS (22).
En otros estudios (9,79,134,141) se ha confirmado que la TVNS en el Holter es benigna en pacientes asintomáticos, mientras que cuando se presenta en pacientes sintomáticos es de mal pronóstico (9). El hallazgo de esta arritmia depende en parte de la duración de la monitorización con Holter. Mulrow y colaboradores (144) evaluaron 198 adultos con Holter de 24, 48, 72, 96, 120 y 144 horas y hallaron que la probabilidad de no detectar episodios de TV disminuye al prolongar el tiempo de registro. Esto indica que se requiere una prolongación del Holter para excluir TV en aquellos pacientes con baja prevalencia de TV.
En dos series prospectivas (53,145) el hallazgo de TV en el Holter tiene una especificidad de 80%, una sensibilidad de 69% y un valor predictivo negativo de 97% como marcador de riesgo de MS cardíaca durante tres años de seguimiento. Pero aunque la correlación entre TV y MS cardíaca ha sido bien demostrada en adultos, el papel de la TVNS como causa de MS sigue siendo objeto de debate. En nuestros resultados la TVNS no fue una variable pronóstica de muerte y tampoco para complicaciones severas en ninguno de los grupos estudiados, obstructivos, no obstructivos, sintomáticos y asintomáticos.
La presencia de 30 o más extrasístoles ventriculares por hora o más de 500 en 24 horas se encuentra en aproximadamente 20% de los casos y se asocia con TV, pero no con la MS (25,53,79).
En la población no terciaria (146), la TVNS registrada en el Holter de 24 horas se asocia con un grado elevado de hipertrofia ventricular y con la severidad de la sintomatología (clase III-IV de la New York Heart Association) (p = 0,04). La TVS ocurre más frecuentemente en pacientes con obstruccción al tracto de salida del ventrículo izquierdo. Para MS, la TVNS en el Holter de 24 horas tiene un valor predictivo negativo y positivo de 95% y 9%, respectivamente, con una sensibilidad y especificidad de 45% y 69%.
Se ha estudiado la importancia de la variabilidad biológica para la evaluación y el manejo de las arritmias ventriculares (147,148). Morganroth y colaboradores (147) hallaron una reducción diaria de la extrasistolia ventricular en 83% en los pacientes con enfermedad coronaria, hipertensión sistémica y miocardiopatía dilatada que atribuyeron al efecto del tratamiento farmacológico y a la propia variabilidad biológica. En los pacientes con MH y TV se ha demostrado (144,149) que se reducen en 61% de los casos por el tratamiento farmacológico más que por la variabilidad biológica.
VARIABLES DERIVADAS DE LA PRUEBA DE ESFUERZO
El 39% de las MS ocurren durante la práctica de ejercicio moderado o severo y se ha encontrado una buena reproducibilidad de arritmias ventriculares severas en las pruebas de esfuerzo realizadas en pacientes con MH (53). La hipotensión relacionada con el ejercicio puede observarse en la prueba de esfuerzo de los pacientes con MH (29,150-153). La importancia potencial de este comportamiento hemodinámico es que un número significativo de las MS cardíacas ocurren durante o después del ejercicio (2,3). Frenneaux y colaboradores (29) estudiaron 129 pacientes (edad media: 41 años) consecutivos y encontraron hipotensión inducida por el ejercicio en 33% de los casos. Esta hipotensión era más común en pacientes jóvenes y se asociaba con historia familiar de MS. La evaluación invasiva demuestra que esta respuesta hemodinámica anormal durante el ejercicio se debe a una respuesta vascular periférica anormal con una exagerada disminución en la resistencia vascular sistémica pese al incremento apropiado del índice cardíaco (29,151).
Sadoul y colaboradores (152) evaluaron consecutiva y prospectivamente una población de pacientes menores de 40 años, con una edad media de 24 años. Se definió una respuesta de presión anormal cuando el incremento de la presión arterial sistólica no era superior a 20 mmHg entre el reposo y el esfuerzo máximo. Se halló una respuesta anormal en 37% de los casos y se asoció con un riesgo aumentado de MS cardíaca independientemente de otros marcadores de riesgo. La hipotensión intraesfuerzo tiene una sensibilidad de 75%, una especificidad de 66%, un valor predictivo negativo de 97% y un valor predictivo positivo de 15% para estratificar el riesgo de MS.
El mecanismo por el cual la respuesta vascular anormal durante el ejercicio se relaciona con la MS no es bien conocido. Yoshida y colaboradores (150) examinaron los estudios gammagráficos de reposo-esfuerzo (201Tl-SPECT) de 105 pacientes y encontraron una respuesta anormal (mayor de 25 mmHg) en 17 (16%) pacientes. Los defectos de perfusión de tipo fijos y reversibles, y la dilatación ventricular izquierda se observaron en 69% de los casos. Los pacientes con respuesta vascular anormal tenían una alta prevalencia de dilatación ventricular. Por lo tanto, concluyeron que la hipotensión inducida en el esfuerzo podría relacionarse con isquemia difusa subendocárdica.
Maki y colaboradores (49) encontraron infradesnivel del segmento ST de tipo isquémico (infradesnivel de tipo horizontal mayor de 0,1 mV a los 0,8 mseg. del punto J) en 31% de los sobrevivientes de MS, en 75% de las muertes cardíacas súbitas durante el ejercicio físico y en 35% de las muertes súbitas no relacionada con el ejercicio. Para otros investigadores (54), los pacientes con una prueba de esfuerzo positiva tenían peor pronóstico, y esto se asociaba con una presión telediastólica ventricular izquierda mayor de 20 mmHg.
En el análisis las pruebas de esfuerzo de los 106 pacientes (56% tratados con betabloqueantes y 23% con verapamilo) de nuestra serie, sólo nueve enfermos presentaron opresión o dolor precordial durante la prueba, en 33% se registró un descenso del segmento ST mayor de 1 mm, y 44% de los enfermos tuvieron una diferencia de presión arterial entre el reposo y el esfuerzo menor a 20 mmHg. Ninguna de las variables analizadas fueron predictivas de mortalidad.
En la MH también se ha descripto un comportamiento hemodinámico anormal después de la ingesta de alimentos debido a un aumento de la frecuencia cardíaca, de la presión auricular derecha, de la presión arterial pulmonar y de la presión capilar pulmonar (154-156). Estos cambios ocurren poco después de ingerir alimentos, con un pico a los 30-60 minutos y persisten por algunas horas (155,156). El incremento del gasto cardíaco después de la ingesta en sujetos normales es debido sobre todo al incremento del volumen diastólico, mientras que en los pacientes con MH sería atribuible al aumento de la frecuencia cardíaca, manteniéndose un volumen sistólico constante (154).
VARIABLES DERIVADAS DEL ECOCARDIOGRAMA DOPPLER
Existen publicaciones en las que se ha estudiado la relación entre los hallazgos ecocardiográficos y el pronóstico de la MH (49,55,157). Candell y colaboradores (55) estudiaron el valor pronóstico de la ecocardiografía en 71 pacientes con MH. Estos enfermos se dividieron en dos grupos según hubieran presentado, o no, uno o varios de los siguientes síntomas o signos: angina, síncope, disnea, IC y FA. Al comparar estadísticamente los resultados de los diferentes parámetros ecocardiográficos entre ambos grupos se llegó a la conclusión de que no había ningún signo que por sí mismo permitiera identificar a los pacientes con peor curso clínico. Sin embargo, los enfermos sintomáticos presentaron un mayor grado de dilatación auricular e hipertrofia ventricular izquierda, si bien ambos parámetros tenían bastantes valores comunes en los dos grupos.
Más recientemente (158), en una serie de 119 pacientes y con seguimiento medio de diez años, observamos en el análisis univariante que las variables predictoras de muerte eran la presencia de FA, el índice auricular izquierdo, el gradiente dinámico y la insuficiencia mitral. Mientras que para complicaciones severas (infarto de miocardio, síncope, angina III-IV y disnea III-IV) la única variable pronóstica fue el índice auricular izquierdo. En las curvas de Kaplan-Meier las variables predictoras de muerte fueron un gradiente dinámico mayor de 50 mmHg (figura 2), un grado de insuficiencia mitral >/= II (figura 3), y un grosor del tabique interventricular >/= 25 mm (figura 4). Para complicaciones severas la única variable pronóstica fue un tamaño de aurícula izquierda mayor de 45 mm y un índice de aurícula izquierda mayor de 25 mm/m2 (figuras 5 y 6). La única variable pronóstica independiente en el análisis multivariable fue la presencia de insuficiencia mitral para mortalidad cardíaca. Nuestro valor de corte para el diámetro auricular izquierdo es semejante al obtenido en el registro italiano (159) de MH con 1.491 pacientes (mayor a 48 mm); constituyendo un predictor de mortalidad tanto para la muerte cardíaca como para la muerte por IC, pero sin asociación con la muerte súbita.
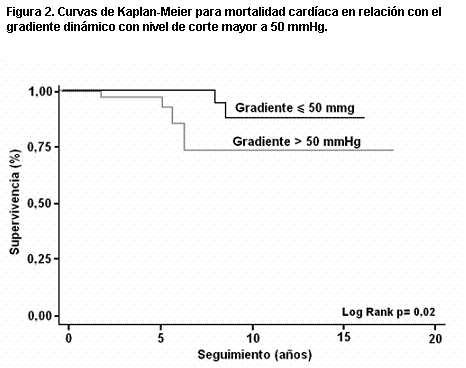
Figura 2. Curvas de Kaplan-Meier para mortalidad cardíaca en relación con el gradiente dinámico con nivel de corte mayor a 50 mmHg.
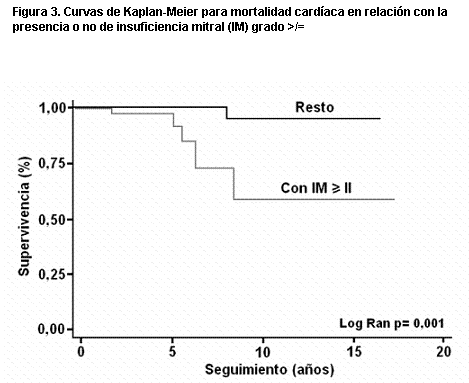
Figura 3. Curvas de Kaplan-Meier para mortalidad cardíaca en relación con la presencia o no de insuficiencia mitral (IM) grado >/= II en el estudio Doppler.
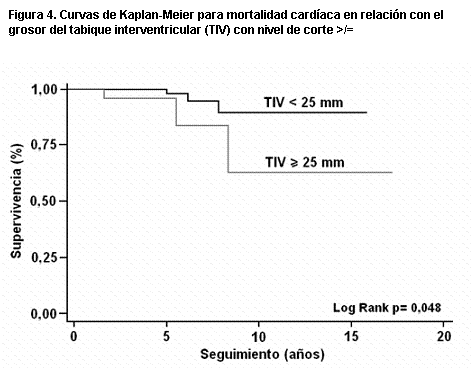
Figura 4. Curvas de Kaplan-Meier para mortalidad cardíaca en relación con el grosor del tabique interventricular (TIV) con nivel de corte >/= 25 mm.
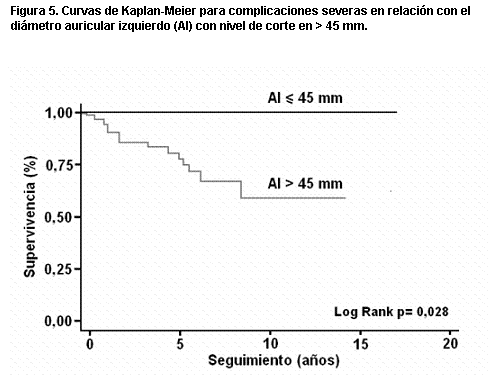
Figura 5. Curvas de Kaplan-Meier para complicaciones severas en relación con el diámetro auricular izquierdo (AI) con nivel de corte en > 45 mm.
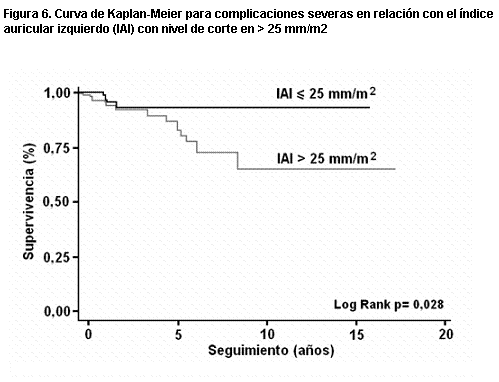
Figura 6. Curva de Kaplan-Meier para complicaciones severas en relación con el índice auricular izquierdo (IAI) con nivel de corte en > 25 mm/m2.
Romeo y colaboradores (38) estudiaron 125 pacientes de forma consecutiva, realizando un seguimiento medio de 7,6 años. En el análisis bivariable los predictores de peor pronóstico fueron la fracción de eyección, la presión arterial pulmonar media, la presión telediastólica del ventrículo izquierdo, las arritmias ventriculares complejas y la regurgitación mitral severa. Utilizando el análisis multivariable los predictores pronósticos independientes fueron: una fracción de eyección reducida y un aumento de la presión arterial pulmonar media. Además se observó que la presencia de gradiente en el tracto de salida del ventrículo izquierdo no tenía significado pronóstico. Esto es discordante con el trabajo de Maki y colaboradores (49), donde, después de analizar 309 pacientes, observaron en el análisis multivariable que la presencia de un gradiente de presión mayor a 30 mmHg en el tracto de salida del ventrículo izquierdo era un predictor independiente de MS (49). Actualmente se ha observado que la importancia pronóstica de la obstrucción dinámica varía en relación con la severidad de los síntomas (160). En los pacientes asintomáticos o levemente sintomáticos la obstrucción es un importante predictor de muerte cardíaca, pero después del desarrollo de sintomatología severa, la clase funcional NYHA es el predictor pronóstico de la MH.
La relación entre la magnitud de la hipertrofia ventricular izquierda y la MS cardíaca no ha sido bien aclarada, pero algunos autores sugieren que la hipertrofia ventricular excesiva puede tener significado pronóstico (1,14,15,57,161,162). Loui y colaboradores (163) analizaron las características morfológicas y clínicas de 34 pacientes con marcada hipertrofia ventricular izquierda (grosores parietales entre 35 y 52 mm, medio: 39 mm) con seguimiento medio de seis años. En este estudio se observó que estos pacientes no tenían peor pronóstico a pesar de tener una masa ventricular tan elevada.
Maron y colaboradores (41) hallaron que la severidad de la hipertrofia no se correlacionaba con el pronóstico, ya que el curso clínico de los pacientes con hipertrofia severa no era necesariamente maligno al compararlos con los pacientes que tenían hipertrofia moderada. En contraste con estos estudios, Spirito y colaboradores (14) compararon 29 pacientes asintomáticos o medianamente sintomáticos que murieron súbitamente o experimentaron paro cardiorrespiratorio por fibrilación ventricular con un grupo control sin antecedentes de MS en estado sintomático similar. El grosor parietal máximo fue significativamente mayor en pacientes que sufrieron MS (26 +/- 7 mm) en comparación al grupo control (21 +/- 5 mm). La hipertrofia ventricular difusa y marcada con un engrosamiento parietal máximo mayor a 30 mm o grosores parietales mayores a 25 mm en dos o más de los cuatro segmentos en los que fue dividido el ventrículo izquierdo fue ocho veces más común en pacientes con MS (38%) en comparación con el grupo control (5%). Una infrecuente localización de la hipertrofia es la apical descripta por primera vez en Japón en 1976 (164). El pronóstico de esta localización se valoró en 26 pacientes durante un seguimiento medio de siete años, encontrándose un curso clínico favorable (0% de mortalidad). Otro tipo poco frecuente de hipertrofia es la hipertrofia asimétrica invertida (de pared posterior, relación TIV/PP menor a 1) que podría ser un marcador ecocardiográfico de peor evolución (165).
La hipertrofia ventricular derecha se observa con relativa frecuencia en las autopsias de los pacientes con MH que fallecen súbitamente, pero su valoración no se realiza sistemáticamente. McKenna y colaboradores (166) evaluaron 73 pacientes con MH mediante ecocardiograma bidimensional, encontrando un grosor parietal de ventrículo derecho que oscilaba entre 4 y 10 mm (medio: 7 mm). El 44% de estos pacientes tenían hipertrofia ventricular derecha que no se asociaba con la presencia de hipertensión arterial pulmonar secundaria, aunque era más común en pacientes con disnea de esfuerzo, hipertrofia ventricular izquierda severa, arritmias supraventriculares y TV en el Holter. El análisis de regresión logística reveló que la presencia de hipertrofia ventricular derecha se asociaba con una enfermedad hipertrófica más severa.
En una minoría de pacientes (10%) con MH existe deterioro progresivo de la función y dilatación ventricular izquierda (167). Después de analizar 139 pacientes se observó tendencia a la dilatación ventricular izquierda en 2% por año asociada a una disminución de la fracción de acortamiento sistólico. Estos pacientes presentaban más disnea, síncope recurrente y un peor pronóstico al compararlos con los enfermos sin dilatación ventricular izquierda (168,169).
La dilatación y disfunción ventricular izquierda es poco frecuente, con una prevalencia de 4,9%. La incidencia de esta alteración en la primera evaluación es de 5,3/1.000 pacientes/año (170). Generalmente son pacientes jóvenes y con historia familiar de MH o MS al compararlos con las MH habituales no dilatadas (171).
VARIABLES DERIVADAS DE TéCNICAS ISOTóPICAS
Tomografía computarizada por emisión de fotón único (SPECT: Single Photon Emission Computed Tomography)
Los defectos de perfusión regionales reversibles no son infrecuentes en los pacientes con MH (172-174) y se han relacionado con arritmias potencialmente letales (64). Que estas alteraciones de la perfusión sean de causa isquémica parece apoyarlo la concordancia existente entre estos defectos y las alteraciones del metabolismo del lactato durante la estimulación eléctrica auricular (175), la desaparición de los defectos tras la miectomía (176), y la excelente concordancia con otros agentes de perfusión (99mTc-MIBI), donde la captación es independiente de la actividad de la bomba de Na-K-ATPasa (177).
Aunque la isquemia miocárdica es una característica de los pacientes adultos con MH, se ha sugerido que también puede jugar un papel importante en pacientes jóvenes asintomáticos (64,172-175) y que podría contribuir al desarrollo del síncope y de la MS. Dilsizian y colaboradores (65), mediante Holter, SPECT de esfuerzo y reposo con 201Tl, y estudio electrofisiológico, sugirieron que en los pacientes jóvenes con MH, el paro cardíaco y el síncope podrían estar relacionados con la isquemia miocárdica. Así pues, la presencia de isquemia en la SPECT de perfusión miocárdica sería un signo predictor de mal pronóstico. El mismo hallazgo se observó posteriormente en los pacientes del grupo de estudio EPIC (Echo Persantine Italian Cooperative) con edades mayores (media 46 años) (178). El grupo de la Clínica Mayo (60) observó que la suma de la puntuación de perfusión de estrés y la suma diferencial de puntuación se asociaban significativamente con la muerte cardíaca. La supervivencia fue significativamente más alta en los pacientes con SPECT normales versus SPECT anormales, sugiriendo que la supervivencia a los cinco años se podría estratificar en categorías de riesgo mediante la suma de la puntuación de perfusión de estrés.
En nuestra población (179) se observó una tendencia a mayor mortalidad en los pacientes con defectos fijos y reversibles, pero sin alcanzar significación estadística. Mientras que para las complicaciones severas (infarto de miocardio, síncope, angina III-IV, disnea III-IV) se alcanzó la significación estadística en los pacientes con defectos fijos (figura 7).
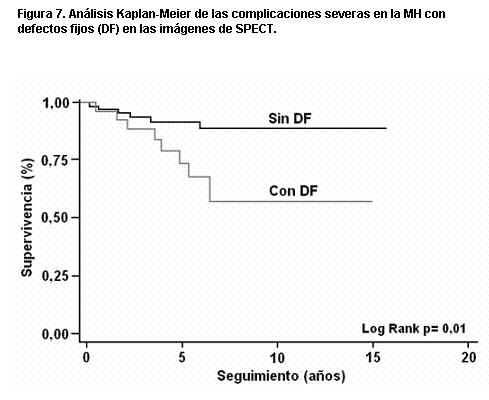
Figura 7. Análisis Kaplan-Meier de las complicaciones severas en la MH con defectos fijos (DF) en las imágenes de SPECT.
Las alteraciones de la perfusión de tipo isquémico (defecto reversible) o de tipo fibrótico o necrótico (defectos fijos) son comunes en los pacientes con MH, ocurriendo más frecuentemente en los pacientes con tabique interventricular de mayor grosor (64). Estas alteraciones en las imágenes gammagráficas tienen una estrecha relación con la TV, la enfermedad del sistema de conducción y la disminución de la fracción de eyección (FE) ventricular izquierda.
Nagata y colaboradores (180) observaron que los pacientes con marcada hipertrofia ventricular izquierda, defectos de perfusión, y enzimas cardíacas elevadas (LDH1 y CPK-MB), tenían una mayor tendencia a desarrollar una miocardiopatía dilatada o disminución de la fracción de eyección, o ambas. Por otro lado, O'gara y colaboradores (172), al estudiar 72 pacientes con 201Tl-SPECT de esfuerzo-reposo y ventriculografía isotópica de equilibrio en reposo, hallaron que 93% de los pacientes con FE menor a 50% tenían defectos fijos o parcialmente reversibles compatibles con cicatriz miocárdica o isquemia severa, mientras que los pacientes con defectos reversibles tenían una FE mayor a 50%. Los defectos fijos en los pacientes con MH son compatibles con descripciones previas de infarto transmural demostrado en autopsias (181). Estos autores también observaron que la disnea y la fatiga fueron síntomas significativamente más comunes en los pacientes con defectos fijos o parcialmente reversibles comparado con el grupo de pacientes con SPECT normal. Los defectos totalmente reversibles ocurrieron predominantemente en pacientes con función sistólica normal o supernormal. Estos autores concluyeron que los defectos fijos o parcialmente reversibles sugieren cicatriz miocárdica o isquemia severa, o ambas, en pacientes con alteración de la función sistólica del ventrículo izquierdo, y que los defectos reversibles pueden reflejar un proceso isquémico.
Posteriormente, Yamada y colaboradores (182) evaluaron la SPECT con talio-201 de 129 pacientes en reposo y luego de administrar 0,56 mg/kg de dipiridamol, encontrando 29% de defectos fijos, 26% de defectos reversibles y 9% con ambos. El defecto fijo fue más común en pacientes con historia de síncope y disnea clase III-IV y mayores diámetros ventriculares. La mayoría de los pacientes con una FE menor a 50% tenían defectos fijos. Los defectos reversibles fueron más comunes en pacientes con historia familiar, mayor diámetro de aurícula izquierda y mayor gradiente dinámico. Por lo tanto, concluyeron que los defectos fijos son frecuentes en pacientes con disnea y deterioro de la función ventricular, pudiendo representar zonas de cicatriz miocárdica, mientras que los defectos reversibles son también comunes aunque no se asocien con isquemia sintomática. Estos hallazgos concuerdan con los resultados de otra publicación en donde los defectos fijos se asocian con síncope, con aumento de las dimensiones del ventrículo izquierdo y reducción de la capacidad al ejercicio (66). También se ha demostrado que 50% de los pacientes tienen un deterioro de la capacidad vasodilatadora (183).
En nuestra experiencia (184), la MH no obstructiva tiene un mayor porcentaje de defectos fijos y los pacientes con defectos fijos tienen una mayor incidencia de ondas Q patológicas en el electrocardiograma (ECG), mayores volúmenes ventriculares, menor FE y un tiempo a la velocidad máxima de llenado ventricular superior. Ninguna variable isotópica fue predictora de mortalidad ni de indicación de marcapaso o miectomía. En un estudio previo en el que analizamos 73 pacientes consecutivos con SPECT, ecocardiograma-Doppler, ventriculografía isotópica y cateterismo cardíaco, con un seguimiento clínico de seis años, no hallamos diferencias significativas entre los pacientes con y sin defectos de perfusión(185).
TOMOGRAFíA POR EMISIóN DE POSITRONES (PET: POSITRON EMISSION TOMOGRAPHY)
Se ha descripto que las imágenes con ácido metil-pentadecanoico (BMIPP) permiten identificar un subgrupo de MH con riesgo elevado de futuras complicaciones cardíacas. Diversos autores (186-190) han observado anormalidades metabólicas en las imágenes de BMIPP en pacientes con MH sin alteraciones de la perfusión y sin alteraciones de la contractilidad. Del mismo modo se ha evidenciado una reducción heterogénea de la captación de ácidos grasos en reposo en pacientes con MH y en otras cardiopatías (191-195). En el análisis de la motilidad parietal regional y en la utilización de imágenes con doble isótopo (201Tl y BMIPP) se ha observado una correlación entre las alteraciones del metabolismo de los ácidos grasos y la discinesia parietal en los pacientes con perfusión miocárdica normal.
Otros radiotrazadores
La captación de anticuerpos antimiosina está aumentada en la fase dilatada de la MH, lo cual sugiere la presencia de cambios necróticos o degeneración de las células miocárdicas, o ambas (196). Estos pacientes desarrollan dilatación ventricular izquierda con hipocontractilidad, pudiendo progresar a la IC (180,197,198). Si bien algunos autores han encontrado estos hallazgos en 100% de los pacientes en fase dilatada (196), los estudios con anticuerpos antimiosina son limitados (196) y, además, en la actualidad no disponemos de este radiofármaco en la práctica.
También en los estudios con meta-yodo- benzil-guanidina (123I-mIBG), un análogo estructural de la adrenalina, las anormalidades de la captación se han relacionado con la presencia de extrasístoles ventriculares en el Holter(199). En un estudio se observó que las anormalidades miocárdicas regionales en la actividad nerviosa simpática pueden hacer sospechar la progresión del daño miocárdico (200). Así pues, las diferencias de los patrones de distribución entre el 123I-mIBG y el 201Tl podrían marcar la progresión de la enfermedad (201).
Recientemente también se ha descripto que la reducción en la densidad de los receptores beta-adrenérgicos en los estudios con C6P-C11 se relaciona con la progresión de la enfermedad (202). La prevalencia de IC varía en relación con los diferentes valores de corte de la relación corazón/mediastino en los estudios de 123I-mIBG. La alteración en la actividad simpática cardíaca es otro predictor de IC(203).
Ventriculografía isotópica en equilibrio
Los parámetros que pueden valorarse con esta exploración son la FE ventricular izquierda y la función diastólica. Las anormalidades de la función diastólica son comunes en estos pacientes (204-207). Se ha observado una prolongación del período isovolumétrico, una disminución de la velocidad máxima de llenado, un aumento del tiempo de llenado y una reducción relativa del volumen de llenado durante el período de llenado rápido con incremento de la contribución auricular (204-208).
No queda claro si estas alteraciones diastólicas tienen importancia pronóstica, pero Newman y colaboradores (209) demostraron que los pacientes que murieron súbitamente tenían reducida la eyección máxima y la velocidad de llenado en el momento del diagnóstico cuando fueron comparados con los sobrevivientes. Esto concuerda con los resultados de Chikamori y colaboradores (210). Estos autores evaluaron 161 pacientes con un seguimiento de tres años y hallaron que los pacientes con MS tenían reducida la velocidad de llenado máxima y mostraban un incremento relativo del volumen de llenado. Hay que tener en cuenta que si bien en el análisis estadístico estos índices se relacionan con la muerte, tienen un valor predictivo positivo bajo. Por lo tanto, la evaluación ventriculográfica de la función diastólica no parece permitir predecir la mortalidad ni contribuir a la identificación de pacientes con elevado riesgo de MS. Pero debe tenerse en cuenta que la ventriculografía isotópica es la técnica más reproducible para el seguimiento de la función sistólica y diastólica ventricular y su respuesta al tratamiento (208).
Nosotros no observamos variables predictivas de mortalidad en los estudios de ventriculografía isotópica en equilibrio, pero sí hallamos una FE significativamente más alta en los pacientes que presentaban síncope.
VARIABLES DERIVADAS DE LA RESONANCIA MAGNéTICA
El aporte pronóstico de la resonancia magnética en estos últimos años se relaciona con el hallazgo de fibrosis miocárdica visualizada en las imágenes de captación tardía de gadolinio (211). La captación tardía de gadolinio se correlaciona directamente con el grosor parietal y con los antecedentes familiares, e inversamente con la FE, con la obstrucción en el tracto de salida del ventrículo izquierdo, con la evidencia de isquemia miocárdica en la prueba de esfuerzo y con un mayor número de factores de riesgo de muerte súbita (212,213). Pero todavía no se sabe si la captación tardía de gadolinio puede predecir la evolución a la IC (214).
VARIABLES DERIVADAS DE LA HISTOLOGíA
No parece que a través de la biopsia endomiocárdica pueda realizarse una estratificación del riesgo en la MH. El parámetro más estudiado hasta la fecha ha sido el desarreglo miofibrillar (139,215). En algunos estudios de pacientes con MH que murieron súbitamente o que sobrevivieron al paro cardíaco se ha encontrado un incremento considerable de la masa ventricular izquierda (14,16,17,216), pero en otros (215) se halló una masa ventricular normal. También se ha visto que la desorganización celular no es patognomónica de la MH y que se la puede encontrar en otras enfermedades (217) o incluso en corazones no patológicos (135,218). El desarreglo miofibrillar está presente siempre aunque no se haya desarrollado aún la hipertrofia ventricular izquierda en pacientes que han presentado MS (215). Esto haría pensar que el desarreglo miofibrillar actuaría como sustrato anatómico histológico para el desarrollo de arritmias malignas y de MS. El desarreglo miofibrillar se asocia, además, con un aumento del tejido conectivo y es cuantitativamente diferente al observado en otras formas de hipertrofia cardíaca (83,219,220). Una de las posibles consecuencias de esta desorganización miofibrillar y del aumento del tejido conectivo es la inestabilidad eléctrica del miocardio, contribuyendo al desarrollo de TVNS y fibrilación ventricular. Además, se piensa que esta misma desorganización miofibrillar es la responsable del desarrollo de fibrosis y dilatación ventricular (167). En el National Heart, Lung and Blood Institute se ha estudiado la distribución y la estructura de la red de colágeno miocárdico en 16 pacientes jóvenes asintomáticos con MH que fallecieron de MS. Se observó una alteración morfológica del colágeno en un área extensa. El aumento del compartimiento de la matriz de colágeno se observó desde edades tempranas con una expansión durante el crecimiento, siendo responsable del incremento del grosor del tabique interventricular. Esto probablemente represente la anormalidad morfológica primaria más importante de la enfermedad (221).
Más recientemente, Varnava y colaboradores (222) estudiaron mediante autopsia los corazones de 75 pacientes fallecidos. Los hallazgos macroscópicos, el porcentaje de desarreglo miofibrillar, el porcentaje de fibrosis, y el porcentaje de enfermedad de pequeño vaso fueron relacionados con el genotipo (estudio de ADN y mutaciones del gen de la troponina T). Observaron que los pacientes con mutaciones de la troponina T tenían un grado severo de desarreglo miofibrillar con moderada hipertrofia y fibrosis. Estos pacientes fallecieron súbitamente y a edades más tempranas, por lo que sugirieron que un desarreglo extenso en ausencia de marcada hipertrofia es el sustrato patológico de la MS en estos pacientes. En contraste con ello, la fibrosis miocárdica sería el sustrato anatómico de la muerte temprana por IC, y además se asocia con un riesgo aumentado de arritmias ventriculares primarias (223).
VARIABLES DERIVADAS DE LA GENéTICA
Hasta el momento se han identificado más de 400 mutaciones diferentes causantes de MH, y en 11 genes que codifican proteínas del sarcómero cardíaco (224): el gen de la cadena pesada de la beta-miosina (B-MHC) (225), el gen de la cadena pesada de la alfa-miosina (224), el gen de la alfa tropomiosina (226), el gen de la troponina cardíaca T (cTnT) (227), el gen de la troponina cardíaca I(228), el gen de la proteína C (MYBPC3) (229), el gen regulador de la cadena ligera de la miosina (230), el gen titin (Arg740Leu) (224), el gen de la alfa-actina cardíaca (231), y más recientemente el gen regulador de la actividad de la AMP-proteinquinasa (PRKAG2) y el gen regulador de la proteína de membrana lisosomal (LAMP-2) (224). Las diferentes manifestaciones clínicas de los pacientes con MH dependen de estas mutaciones. Se han publicado diferentes análisis de genotipo y fenotipo. Las mutaciones del gen B-MHC se asocian con una marcada hipertrofia ventricular izquierda (26,232) en contraste con las mutaciones del gen cTnT, que se acompañan de una hipertrofia de grado leve a moderado (233).
Algunas mutaciones en el gen de la cadena pesada de la miosina cursan con peor pronóstico (26,232), mientras que seis mutaciones en el gen de la cTnT se han relacionado con una elevada incidencia de MS (227,231,233,234).
Parece ser que tanto las mutaciones de la cadena pesada de la -miosina con hipertrofia significativa y con una penetrancia cercana al 95%(235), como las mutaciones de la troponina T, presentes en 15% de las familias estudiadas, con una hipertrofia leve y de baja penetrancia (25%), se asocian con un mal pronóstico (227,233). Moolman y colaboradores (227) enfatizan la importancia del 'screening' de las mutaciones del gen cTnT. Fujino y colaboradores (236) hallaron que la sustitución Arg92Trp en el gen de la troponina T cardíaca tiene alto grado de penetrancia, moderada hipertrofia y una progresión temprana a la miocardiopatía dilatada en pacientes japoneses. Pero recientemente (237) se ha observado que no todas estas mutaciones predicen una evolución desfavorable de la enfermedad. Así, la primera mutación del gen cTnT cursaría con un pronóstico benigno al igual que la mutación en el exón 25 del gen MyBP-C con penetrancia incompleta y dependiente de la edad (238). En estos últimos años la investigación genética ha aportado datos pronósticos importantes de los pacientes con MH, pero queda por demostrar que en el futuro próximo sea un estudio fundamental en la estratificación del riesgo en la práctica clínica (239,240).
RESUMEN
La ausencia de factores de riesgo identifica baja probabilidad de muerte y la presencia de dos o más factores de riesgo (presentes en 12% de los pacientes) ocasionan un aumento acumulativo del riesgo asociándose con 4% a 5% anual de riesgo de muerte (241). El cambio que el tratamiento pueda representar en la evolución de la MH es otro de los aspectos controvertidos. Parece haber evidencia clínica de que la terapia con cardiodesfibrilador implantable puede prevenir la MS en los pacientes con MH y múltiples factores de alto riesgo, mientras que las recomendaciones terapéuticas para pacientes con un solo factor de riesgo continúan siendo controvertidas.
Puede afirmarse que realizar una estratificación del riesgo y valorar el pronóstico con exactitud en cada paciente con MH es muy difícil. Las publicaciones encaminadas a analizar diferentes variables pronósticas mediante diversas exploraciones están sesgadas por los criterios de inclusión de los enfermos (edad, población ambulatoria u hospitalaria), tiempo de seguimiento, y nivel de complejidad de las técnicas empleadas. En las publicaciones que incluyen pacientes de mediana edad asintomáticos, estudiados ambulatoriamente y con un seguimiento no superior a los tres años, el pronóstico suele ser bueno, pero en los pacientes jóvenes sintomáticos, estudiados en centros hospitalarios de tercer nivel y con un seguimiento superior a cinco años, la mortalidad anual es más elevada. En estos últimos años se han descripto numerosas variables predictoras de mal pronóstico que el cardiólogo debe saber valorar adecuadamente cuando se enfrenta a un paciente determinado. La mayoría de estas variables son fácilmente identificables con una correcta anamnesis (síncope repetitivo en el joven, historia familiar maligna de MS, antecedente personal de parada cardiorrespiratoria por fibrilación ventricular, TV sostenida, disnea y angina de grado IV), una buena exploración del paciente (arritmia completa, soplo sistólico) y la práctica de técnicas de rutina como son el ECG (FA), el Holter (episodios de TVNS, sobre todo cuando son múltiples, repetitivos o prolongados), la prueba de esfuerzo (respuesta plana o hipotensiva de la presión arterial) y el ecocardiograma Doppler (hipertrofia ventricular extrema >/= 30 mm, insuficiencia mitral, gradiente dinámico). De todas éstas, las variables de mayor riesgo de MS aceptadas actualmente para el implante de un cardioversor-desfibrilador implantable son la fibrilación ventricular, la taquicardia ventricular sostenida espontánea, la historia familiar de MS cardíaca, el síncope inexplicado, el grosor parietal ventricular izquierdo >/= 30 mm, la respuesta anormal de la presión arterial durante el ejercicio y la TVNS espontánea. Las variables isotópicas (necrosis, isquemia, denervación autonómica), de la resonancia magnética (necrosis) y genéticas (mutaciones), parecen tener un valor pronóstico secundario en el momento actual.
BIBLIOGRAFíA
1. McKenna WJ, Camm AJ. Sudden death in hypertrophic cardiomyopathy. Assessment of patients at high risk. Circulation 1989; 80: 1489-92.
2. Maron BJ, Roberts WC, Epstein SE. Sudden death in hypertrophic cardiomyopathy: a profile of 78 patients. Circulation 1982, 65: 1388-94.
3. Maron BJ, Roberts WC, McAllister HA, Rosing DR, Epstein SE. Sudden death in young athletes. Circulation 1980; 62: 218-29
4. Spirito P, Chiarella F, Carratino L, Zoni Berisso M, Bellotti P, Vecchio Carlo. Clinical course and prognosis of hypertrophic cardiomyopathy in an outpatient population. N Engl J Med 1989; 320: 749-55.
5. Shapiro LM, Zezulka A. Hypertrophic Cardiomyopathy: A Common disease with a good prognosis: Five year experience of a district general hospital. Br Heart J 1983; 50: 530-3.
6. Maron BJ, Nichols PF, Pickle LW, Wesley YE, Mulvihill JJ. Patterns of inheritance in hypertrophic cardiomyopathy. Assessment by M-mode and two-dimensional echocardiography. Am J Cardiol 1984; 53: 1087-94.
7. Maron BJ, Spirito P. Impact of patient selection biases on the perception of hypertrophic cardiomyopathy and its natural history. Am J Cardiol 1993; 72: 970-2.
8. Maron BJ, Mathenge R, Casey SA, Poliac LC, Longe TF. Clinical profile of hypertrophic cardiomyopathy identified de novo in rural communities. J Am Coll Cardiol 1999; 33: 1590-5.
9. Fananapazir L, Chang AC, Epstein SE, McAreavey D. Prognostic determinants in hypertrophic cardiomyopathy. Prospective evaluation of a therapeutic strategy based on clinical, Holter, hemodynamic, and electrophysiological findings. Circulation 1992; 86: 730-40.
10. Cecchi F, Maron BJ, Epstein SE. Long-term outcome of patients with hypertrophic cardiomyopathy successfully resuscitated after cardiac arrest. J Am Coll Cardiol 1989; 13: 1283-8.
11. Maron BJ, Lipson LC, Roberts WC, Savage DD, Epstein SE. Malignant hypertrophic cardiomyopathy: identification of a subgroup of families with unusually frequent premature death. Am J Cardiol 1978; 41: 1133-40.
12. Fananapazir L, Epstein SE. Hemodynamic and electrophysiologic evaluation of patients with hypertrophic cardiomyopahty surviving cardiac arest. Am J Cardiol 1991; 67: 280-7.
13. Gómez-Marino MA, Alonso Rodríguez C, Méndez Martín-Allegue A. Miocardiopatía hipertrófica con ondas T isoeléctricas. Rev Esp Cardiol 1996; 49: 567-79.
14. Spirito P, Maron BJ. Relation between extent of left ventricular hypertrophy and ocurrence of sudden cardiac death in hypertrophic cardiomyopathy. J Am Coll Cardiol 1990, 15: 1521-6.
15. Hecht GM, Panza JA, Maron BJ. Clinical course of middle-aged asymtomatic patients with hypertrophic cardiomyopathy. Am J Cardiol 1992; 69: 935-40.
16. Maron BJ, Bonow RO, Cannon RO 3rd, Leon MB, Epstein SE. Hypertrophic cardiomyopathy. Interrelations of clinical manifestations, pathophysiology, and therapy. N Engl J Med 1987; 316: 780-9.
17. Wigle ED, Sasson Z, Henderson MA, Ruddy TD, Fulop J, Rakowski H, et al. Hypertrophic cardiomyopathy: The importance of the side and entent of hypertrophy: A review. Prog Cardiovasc Dis 1985; 28: 1-83.
18. Hecht GM, Klues HG, Roberts WC, Maron BJ. Coexistence of sudden cardiac death and end-stage heart failure in familial hypertrophic cardiomyopathy. J Am Coll Cardiol 1993; 22: 489-97.
19. Nicod P, Polikar R, Peterson KL. Hypertrophic cardiomyopathy and sudden death. N Engl J Med 1988; 318: 1255-7.
20. Maron BJ, Kogan J, Proschan MA, Hecht GM, Roberts WC. Circadian variability in the occurrence of sudden cardiac death in patients with hypertrophic cardiomyopathy. J Am Coll Cardioll 1994; 23: 1405-9.
21. McKenna WJ, Deanfield JE. Hypertrophic cardiomyopathy: an important cause of sudden death. Arch Dis Child 1984, 59: 971-5.
22. Maron BJ, Henry Wl, Clark CE, Redwood DR, Roberts WC, Epstein SE. Asymmetric septal hypertrophy in childhood. Circulation 1976; 53: 9-19.
23. Maron BJ, Roberts WC, Edwards JE, McAllister HA, Foley DD, Epstein SE. Sudden death in patients with hypertrophic cardiomyopathy: characterization of 26 patients without functional limitation. Am J Cardiol 1978; 41: 803-10.
24. Maron BJ, Fananapazir L. Sudden cardiac death in hypertrophic cardiomyopathy. Circulation 1992; 85 (1 Suppl): I 157-63.
25. McKenna WJ, England D, Doi YL, Deanfield JE, Oakley C, Goodwin JF. Arrhythmia in hypertrophic cardiomyopathy. I: influence on prognosis. Br Heart J 1981, 46: 168-72.
26. Watkins H, Rosenzweig A, Hwang DS, Levi T, McKenna W, Seidman CE, et al. Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N Engl J Med 1992; 326: 1108-14.
27. Spirito P, Watson RM, Maron BJ. Relation between extent of left ventricular hypertrophy and occurrence of ventricular tachycardia in hypertrophic cardiomyopathy. Am J Cardiol 1987; 60: 1137-42.
28. McKenna W, Deanfield J, Faruqui A, England D, Oakley C, Goodwin J. Prognosis in hypertrophic cardiomyopathy: role of age and clinical, electrocardiographic and hemodynamic features. Am J Cardiol 1981; 47: 532-8.
29. Frenneaux MP, Counihan PJ, Caforio ALP, Chikamori T, McKenna WJ. Abnormal blood pressure response during exercise in hypertrophic cardiomyopathy. Circulation 1990; 82: 1995-2002.
30. Maron BJ, Cecchi F, McKenna WJ. Risk factors and stratification for sudden cardiac death in patients with hypertrophic cardiomyopathy. Br Heart J 1994; 72: S13-S18.
31. Kofflar MJ, Waldstein DJ, Vos J, Ten Cate FJ. Prognosis in hypertrophic cardiomyopathy observed in a large clinic population. Am J Cardiol 1993; 72: 939-43.
32. Candell-Riera J, Romero-Farina G, Galve E, Armadans L, Palet J, Castell-Conesa J. Hypertrophic cardiomyopathy in adult patients. Clinical course and prognostic factors in 119 patients. Med Clin (Barc)2004; 123: 1-4.
33. Brugada J. Muerte súbita en la miocardiopatía hipertrófica. Rev Esp Cardiol 1998; 51: 991-6.
34. Wigle ED, Rakowski H, Kimball BP, Williams WG. Hypertrophic cardiomyopathy: clinical spectrum and treatment. Circulation 1995; 92: 1680-92.
35. Maron BJ, Shirani J, Poliac LC, Mathenge R, Roberts WC, Mueller FO. Sudden death in young competitive athletes: clinical, demographic and pathological profiles. JAMA 1996; 276: 199.
36. Hardarson T, de la Calzada CS, Curiel R, Goodwin JF. Prognosis and mortality of hypertrophic obstructive cardiomyopathy. Lancet 1973; 2: 1462-7.
37. Shah PM, Adelman AG, Wigle ED, Gobel FL, Burchell HB, Hardarson T, et al. The natural (and unnatural) history of hypertrophic obstructive cardiomyopathy. Circ Res 1974; 35: suppl II: 179-95.
38. Romeo F, Pelliccia F, Cristofani R, Martuscelli E, Reale A. Hypertrophic cardiomyopathy: is a left ventricular outflow tract gradient a major prognostic determinant? Eur Heart J 1990; 11: 233-40.
39. Cecchi F, Olivotto I, Montereggi A, Santoro G, Dolara A, Maron BJ. Hypertrophic cardiomyopathy in Tuscany: clinical course and outcome in an unselected regional population. J Am Coll Cardiol 1995; 26: 1529-36.
40. Kyriakidis M, Triposkiadis F, Anastasakis A, Theopistou A, Tocta R, Barbetseas J, et al. Hypertrophic cardiomyopathy in Greece: clinical course and outcome. Chest 1998; 114: 1091-6.
41. Maron BJ, Casey SA, Poliac LC, Gohman TE, Almquist AK, Aeppli DM. Clinical course of hypertrophic cardiomyopathy in a regional united states cohort. JAMA 1999; 281: 2288.
42. Takagi E, Yamakado T, Nakano T. Prognosis of completely asymptomatic adult patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999; 33: 206-11.
43. Ikeda H, Maki S, Yoshida N, Murohara T, Adachi H, Koga Y, Imaizumi T. Predictors of death from congestive heart failer in hypertrophic cardiomyopathy. Am J Cardiol 1999; 83: 1280-3
44. Spirito P, Bellone P, Harris KM, Bernabò P, Bruzzi P, Maron BJ. Magnitud of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med 2000; 342: 1778-85.
45. Maron BJ, Olivotto I, Spirito P, Casey SA, Bellone P, Gohman TE, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000; 102: 858-64.
46. Elliott PM, Sharma S, Varnava A, Poloniecki J, Rowland E, McKenna WJ. Survival after cardiac arrest or sustained ventricular tachycardia in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999; 33: 1596-601.
47. Slade AKB, Camm AJ. Risk assessment and prevention of sudden cardiac death in hypertrophic cardiomyopathy. Arch Mal Couer 1996; 89 (I): 37.
48. Maron BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, et al. American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents; European Society of Cardiology Committee for Practice Guidelines. American College of Cardiology/European Society of Cardiology Clinical Expert Consensus Document on Hypertrophic Cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. Eur Heart J 2003; 24: 1965-91.
49. Maki S, Ikeda H, Muro A, Yoshida N, Shibata A, Koga Y, et al. Predictors of sudden cardiac death in hypertrophic cardiomyopathy. Am J Cardiol 1998; 82: 774-8.
50. Cannan CR, Reeder GS, Bailey KR, Melton III LJ, Gersh BJ. Natural history of hypertrophic cardiomyopathy. A population-based study, 1976 through 1990. Circulation 1995; 92: 2488-95.
51. Efthimiadis GK, Giannakoulas G, Parharidou DG, Karvounis HI, Mochlas ST, Styliadis IH, et al. Prevalence of systolic impairment in an unselected regional population with hypertrophic cardiomyopathy. Am J Cardiol 2006; 98: 1269-72.
52. Frank S, Braunwald E. Idiopathic hypertrophic subaortic stenosis. Clinical analysis of 126 patients with emphasis on the natural history. Circulation 1968; 37: 759-88.
53. Maron BJ, Savage DD, Wolfson JK, Epstein SE. Prognostic significance of 24 hour ambulatory electrocardiographic monitoring in patients with hypertrophic cardiomyopathy: a prospective study. Am J Cardiol 1981; 48: 252-7.
54. Koga Y, Itaya K, Toshima H. Prognosis in hypertrophic cardiomyopathy. Am Heart J 1984; 108: 351-9.
55. Álvarez Auñón A, Candell-Riera J, Caravedo Balda F, García del Castillo H, Soler Soler J. ¿Existen diferencias ecocardiográficas entre los pacientes sintomáticos y asintomáticos afectos de miocardiopatía hipertrófica? Rev Esp Cardiol 1988; 41: 143-147.
56. Fay WP, Taliercio ChP, Ilstrup DM, Tajik AJ, Gersh BJ. Natural history of hypertrophic cardiomyopathy in the elderly. J Am Coll Cardiol 1990; 16: 821-6.
57. Aron LA, Hertzeanu HL, Fisman EZ, Nosrati LL, Kellermann JJ. Prognosis of nonobstructive hypertrophic cardiomyopathy. Am J Cardiol 1991; 15: 215-7.
58. Spirito P, Bellone P. Natural history of hypertrophic cardiomyopathy. Br Heart J 1994; 72: S10-S12.
59. Nasermoaddeli A, Miura K, Matsumori A, Soyama Y, Morikawa Y, Kitabatake A, Inaba Y, et al. Prognosis and prognostic factors in patients with hypertrophic cardiomyopathy in Japan: results from a nationwide study. Heart 2006; [Epub ahead of print].
60. Sorajja P, Chareonthaitawee P, Ommen SR, Miller TD, Hodge DO, Gibbons RJ. Prognostic utility of single-photon emission computed tomography in adult patients with hypertrophic cardiomyopathy. Am Heart J 2006; 151: 426-35.
61. Maron BJ. Hypertrophic cardiomyopathy. Curr Probl Cardiol 1993; 18: 639-704.
62. McKenna WJ, Goodwin JF. The natural history of hypertrophic cardiomyopathy. Current Problems in Cardiology 1981; 20: 1-26.
63. Ly HQ, Greiss I, Talakic M, Guerra PG, Macle L, Thibault B, et al. Sudden death and hypertrophic cardiomyopathy: a review. Can J Cardiol 2005; 21: 441-8.
64. Von Dohlen TW, Prisant LM, Frank MJ. Significance of positive or negative thallium-201 scintigraphy in hypertrophic cardiomyopathy. Am J Cardiol 1989; 64: 498-503.
65. Dilsizian V, Bonow RO, Epstein SE, Fananapazir L. Myocardial ischemia detected by thallium scintigraphy is frequently related to cardiac arrest and syncope in young patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 1993; 22: 796-804.
66. Yamada M, Elliot PM, Kaski JC, Prasad K, Gane JN, Lowe CM, et al. Dipyridamole stress thallium-201 perfusion abnormalities in patients with hypertrophic cardiomyopathy. Relationship to clinical presentation and outcome. Eur Heart J 1998; 19: 500-7.
67. Vassalli G, Seiler Ch, Hess O. Risk stratification in hypertrophic cardiomyopathy. Current Opinion in Cardiology 1994; 9: 330-6.
68. Watson RM, Schwatrz JL, Baron BJ, Tucker E, Rosing DR, Josephson ME. Inducible polymorphic ventricular tachycardia and ventricular fibrillation in a subgroup of patients with hypertrophic cardiomyooathy at high risk for sudden death. J Am Coll Cardiol 1987; 10: 761-74.
69. Maron BJ, Gottdiener JS, Epstein SE. Patterns and significance of the distribution of left ventricular hypertrophy in hypertrophic caardiomyopaathy: A wide angle, two-dimensional echocardiographic study of 125 patients. Am J Cardiol 1981; 48: 418-28.
70. Hada Y, Sakamoto T, Amano K, Yamaguchi T, Takenaka K, Takahashi H, et al. Prevalence of hypertrophic cardiomyopathy in a population of adult Japanese workers as detected by echocardiographic screening. Am J Cardiol 1987; 59: 183-4.
71. McKenna WJ, Sharma S, Elliot PM. Management strategies in hypertrophic cardiomyoapthy: influence of age and morphology. Eur Heart J 2001; 3(Suppl L): L10-L14.
72. Henry WL, Clarke CE, Epstein SE. Asymmetrical septal hypertrophic (ASH): the unifying link in the IHSS disease spectrum. Observations regarding its pathogenesis, pathophysiology and course. Circulation 1973; 47: 827-32.
73. Savage DD, Castelli WP, Abbott RD. Hypertrophic cardiomyopathy and its markers in the general population: The great masquerader revisited: The Framinghan Study. J Cardiovasc Ultrason 1983; 2: 41.
74. Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation 1995; 92: 785-9.
75. Abbassi AS, MacAlpin RN, Erb LM, Pearce ML. Echocardiographic diagnosis of idiopathic hypertrophic cardiomyopathy with outflow obstruction. Circulation 1972; 46: 897-904.
76. Liouville H. Retrecissement cardiaque sous aortique. Gaz Med Paris 1869; 24: 161-4.
77. Maron BJ. The giant negative T wave revisited... in hypertrophic cardiomyopathy. J Am Coll Cardiol 1990; 15: 972-3.
78. Bunch TJ, Chandrasekaran K, Ehrsam JE, Hammill SC, Urban LH, Hodge DO, et al. Prognostic significance of exercise induced arrhythmias and echocardiographic variables in hypertrophic cardiomyopathy. Am J Cardiol 2007; 99: 835-8.
79. Savage DD, Seids SF, Maron BJ, Myers AJ, Epstein SE. Prevalence of arrhythmias during 24-hour electrocardiographic monitoring and exercice testing in patients with obstructive and nonobstructive hypertrophic cardiomyopathy. Circulation 1979, 59: 866-75.
80. Glancy DL, O'Brien KP, Gold HK, Epstein SE. Atrial fibrillation in patients with idiopathic hypertrophic subaortic stenosis. Br Heart J 1970: 32: 652-9.
81. Henry WL, Morganroth J, Pearlman AS, Clark CE, Redwood DR, Itscoitz SB, Epstein SE. Relation between echocardiographically determined left atrial size and atrial fibrillation. Circulation 1976; 53: 273-9.
82. Watson DC, Henry WL, Epstein SE. Effects of operation on left atrial size and the occurence of atrail fibrillation in patients with hypertrophic subaortic stenosis. Circulation 1977; 55: 178-81.
83. Braunwald E, Lambrew CT, Rockoff D, Ross J, Morrow AG. Idiopathic hypertrophic subaortic stenosis: a description of the disease based upon an analysis of 64 patients. Circulation 1964; 29 (suppl IV): 3-119.
84. Krikler DM, Davies MJ, Rowland E, Goodwin JF, Evans RC, Shaw DB. Sudden death in hypertrophic cardiomyopathy: associated accessory atrioventricular pathways. Br Heart J 1980; 43: 245-51.
85. Akasaka K, Kawashima E, Shiokoshi T, Ishii Y, Hasebe N, Kikuchi K. Left atrial function and left atrial appendage flow velocity in hypertrophic cardiomyopathy: comparison of patients with and without paroxysmal atrial fibrillation. J Cardiol 1998; 32: 31-8.
86. Stafford WJ, Trohman RG, Bilsker M, Zaman L, Castellanos A, Myerburg RJ. Cardiac arrest in an adolescent with atrial fibrillation and hypertrophic cardiomyopathy. J Am Coll Cardiol 1986; 7: 701-4.
87. Robinson K, Frenneaux MP, Stockins B, Karatasakis G, Poloniecki JD, McKenna WJ. Atrial fibrillation in hypertrophic cardiomyopathy: a longitudinal study. J Am Coll Cardiol 1990; 15: 1279-85.
88. Greenspan AM. Hypertrophic cardiomyopathy and atrial fibrillation: a change of perspective. J Am Coll Cardiol 1990; 15: 1286-7.
89. Kuchar Dl, Thornburn CW, Sammel NL. Prediction of serious arrhythmic events after myocardial infarction: signal-averaged electrocardiogram, Holter monitoring and radionuclide ventriculogram. J Am Coll Cardiol 1987; 9: 531-8.
90. Gomes JA, Winters Sl, Steward D, Horowitz S, Milner M, Barreca P. A new noninvasive index to predict sustained ventricular tachycardia and sudden death in the first year after myocardial infarction based on the signal-averaged electrocardiogram, radionuclide ejection fraction and Holter monitoring. J Am Coll Cardiol 1987,10: 349-57.
91. Keeling PJ, Kulakowski P, Yi G, Slade AKB, Bent SE, McKenna WJ. Usefulness of signal-averaged electrocardiogram in idiopathic dilated cardiomyopathy for identifying patients with ventricular arrhythmias. Am J Cardiol 1993; 72: 78-84.
92. Turitto G, Ahuja RK, Bekheit S, Caref EB, Ibrahim B, El-Sherif N. Incidence and prediction of induced ventricular tachyarrythmias in idiopathic dilated cardiomyopathy. Am J Cardiol 1994; 73: 770-3.
93. Kulakowski P, Counihan PJ, Camm AJ, McKenna WJ. The value of time and frequency domain, and spectral temporal mapping analysis of the signal-averaged electrocardiogram in identification of patients with hypertrophic cardiomyopathy at increased risk of sudden death. Eur Heart J 1993; 14: 941-50.
94. Chang AC, McAreavey D, Fananapazir L. Identification of patients with hypertrophic cardiomyopathy at high risk for sudden death. Curr Opin Cardiol 1995; 10: 9-15.
95. Fananapazir L, Tracy CM, Leon, Winkler JB, Cannon RO 3rd, Bonow RO, et al. Electrophysiologic abnormalities in patients with hypertrophic cardiomyopathy. A consecutive analysis in 155 patients. Circulation 1989; 80: 1259-68.
96. Maron BJ, Epstein SE, Roberts WC. Causes of sudden death in competitive athletes. J Am Coll Cardiol 1986; 7: 204-14.
97. Fananapazir L, Maron BJ. Arrhythmias in hypertrophic cardiomyopathy. In Current management of arrhythmias. Edited by Horowitz L. Philadelphia: BC Decker, Inc: 1990: p182.
98. Schwartz PJ. Idiopathic long QT syndrome: progress and questions. Am Heart J 1985; 109: 399-411.
99. Jackman WM, Friday KJ, Anderson JL, Aliot EM, Clark M, Lazzara R. The long QT syndromes: a critical review, new clinical observations and a unifying hypothesis. Prog Cardiovasc Dis 1988; 31: 115-72.
100. Day CP, McComb JM, Campbell RWF. QT dispersion: an indication of arrhythmia risk in patients with long QT intervals. Br Heart J 1990; 63: 342-4.
101. Hii JTY, Wyse G, Gillis AM, Duff HJ, Solylo MA, Mitchell B. Precordial QT dispersion as a marker of torsade de pointes. Disparate effects of class Ia antiarrhythmic drugs and amiodarone. Circulation 1992; 86: 1376-82.
102. De Ambroggi L, Negroni MS, Monza E, Bertoni T, Schwarz PJ. Dispersion of ventricular repolarization in the long QT syndrome. Am J Cardiol 1991; 68: 614-20.
103. Linker NJ, Colonna P, Kekwick CA, Till J, Camm JA, Ward DE. Assessment of dispersion in symptomatic patients with congenital long QT syndromes. Am J Cardiol 1992; 69: 634-8.
104. Day CP, McComb JM, Matthews J, Campbell RWF. Reduction in QT dispersion by sotalol following myocardial infarction. Eur Heart J 1991; 12: 423-7.
105. Dritsas A, Gilligan D, Nihoyannopoulos P, Oakley CM. Amiodarone reduces QT dispersion in patients with hypertrophic cardiomypathy. Int J Cardiol 1992; 36: 345-9.
106. Mirvis DM. Spatial variation of QT intervals in normal persons and patients with acute myocardial infarction. J Am Coll Cardiol 1985; 5: 625-31.
107. Engler RL, Smith P, LeWinter M, Gosink B, Johnson A. The electrocardiogram in asymmetric septal hypertropy. Chest 1979; 75: 167-73.
108. Dristas A, Sbarouni E, Gilligan D, Nihoyannopoulos P, Oakley CM. Qt-interval abnormalities in hypertrophic cardiomyopathy. Clin Cardiol 1992; 15: 739-42.
109. Buja G, Miorelli M, Turrini P, Melacini P, Nava A. A comparison of QT dispersion in hypertrophic cardiomyopathy between patients with and without ventricular arrhythmia and sudden death. Am J Cardiol 1993; 72: 973-6.
110. Martin AB, Garson A, Perry JC. Prolonged QT interval in hypertrophic and dilated cardiomyopathy in children. Am Heart J 1994; 127: 64-70.
111. Doevendans PA. Hypertrophic cardiomyopathy. Do we have the algorihm for life and death? Circulation 2000; 101: 1242-6.
112. Atiga Wl, Fananapazir L, McAreavey, Calkins H, Berger RD. Temporal repolarization lability in hypertrophic cardiomyoapthy caused by beta-myosin heavy-chain gene mutations. Circulation 2000, 101: 1237-42.
113. Yi G, Poloniecki J, Dickie S, Elliott PM, Malik M, McKenna WJ. Is QT dispersion associated with sudden cardiac death in patients with hypertrophic cardiomyopathy? Ann Noninvasive Electrocardiol 2001; 6: 209-15.
114. Berger RD, Kasper EK, Baughman KL, Marban E, Calkins H, Tomaselli GF. Beat-to-beat QT interval variability. Novel evidence for repolarization labality in ischemic and nonischemic dilated cardiomyopathy. Circualtion 1997; 96: 1557-65.
115. Zipes DP, Miyazaki T. The autonomic nervous system and the heart: basis for understanding interactions and effects on arrhythmia development. In: Zipes DP, Jalife J, editores. Cardiac Electrophysiology: From Cell Bedside. Philadelphia: WB Saunders, 1990: p312.
116. Kleiger RE, Miller JP, Bibber JT, Moos AJ. Decreased heart rate variability and its association with increased mortality after acute myocardial infarction. Am J Cardiol 1987; 59: 256-62.
117. Pagani M, Lombardi F, Guzzetti S, Rimoldi O, Furlan R, Pizzinelli P, et al. Power spectral analysis of heart rate and arterial pressure variabilities as a marker of sympatho-vagal interaction in man and conscious dog. Circ Res 1986; 59: 178-93.
118. Lombardi F, Torzillo D, Sandrome G, Dalla Vecchia L, Finocchiaro ML, Bernasconi R, et al. Beta-blocking effect of propafenone based on spectral analysis of heart rate variability. Am J Cardiol 1992; 70: 1028-34.
119. Malliani A, Lombardi F, Pagani M. Power spectrum analysis of heart rate variability a tool to explore neural regulatory mechanisms. Br Heart J 1994; 71: 1-2.
120. Arribas Jiménez A, Martín Luengo C, Sáez Jiménez A, Sánchez L, Fernández P, Pabn Osuna P, et al. Aproximación al conocimiento del estado del sistema nervioso autonómico en la miocardiopatía hipertrófica mediante el análisis de la variabilidad de la frecuencia cardiaca. Rev Esp Cardiol 1998; 51: 286-91.
121. Fei L, Slade A, Prasad K, Malik M, McKenna W, John Camm A. Is there increased sympathetic activity in patients with hypertrophic cardiomyopathy? J Am Coll Cardiol 1995; 26: 472-80.
122. Ajiki K, Murakawa Y, Yanagisawa-Miwa A, Usui M, Yamashita T, Oikawa N, Inoue H. Autonomic nervous system activity in idiopathic dilated cardiomyopathy and in hypertrophic cardiomyopathy. Am J Cardiol 1993; 71: 1316-20.
123. Gilligan DM, Chan Wl, Sbarouni E, Nihoyannopoulos P, Oakley CM. Autonomic function in hypertrophic cardiomyopathy. Br Heart J 1993; 69: 525-9.
124. Louie EK, Edwards LC III. Hypertrophic cardiomyopathy. Prog Cardiovasc Dis 1994; 35: 275-308.
125. Zipes DF. Influences of myocardial ischemia and infarction on autonomic innervation of heart. Circulation 1990; 82: 1095-105.
126. Grassi G, Giannattasio C, Seravalle G, Cattaneo BM, Cléroux J, Mancia G. Cardiogenic reflex and left ventricular hypertrophic. Eur Heart J 1990; 11(Supl G): 95-9.
127. Kawasaki T, Azuma A, Sakatani T, Hadase M, Kamitani T, Kawasaki S, et al. Prognostic value of heart rate variability in patients with hypertrophic cardiomyopathy. J Electrocardiol 2003; 36: 333-8.
128. Mason JW, Winkle RA. Accuracy of the ventricular tachycardia-induction study for predicting longterm efficacy and inefficacy of antiarrhythmic drugs. N Engl J Med 1980; 303: 10737.
129. Richards DA, Cody DV, Dennis AR, Russell PA, Young AA, Uther JB. Ventricular electrical instability: a predictor of death after myocardial infarction. Am J Cardiol 1983; 51: 75-80.
130. Ruskin JN, DiMarco JP, Garan H. Out-of- hospital cardiac arrest: electrophysiologic observations and selection of long-term antiarrhythmic therapy. N Engl J Med 1980; 303: 607-13.
131. Lembo NJ, Dell-Italia LJ, Crawford MH, O'Rourke RA. Bedside diagnosis of systolic murmurs. N Engl J Med 1988, 318: 1572-8.
132. Geibel A, Brugada P, Zehender M, Stevenson W, Waldecker B, Wellens HJ. Value of programmed electrical stimulation using a standardized ventricular stimulation protocol in hypertrophic cardiomyopathy. Am J Cardiol 1987; 60: 738-9
133. Kuck KH, Kunze KP, Schlüter M, Nienaber CA, Costard A. Programmed electrical stimulation in hypertrophic cardiomyopathy. Results in patients with and without cardiac arrest or syncope. Eur Heart J 1988; 9: 177-85.
134. Ariga WL, McAreavey D, Epstein ND. Risk stratification of asymptomatic patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 1994; 23: 110A.
135. Davis MJ. The current status of myocardial disarray in hypertrophic cardiomyopathy. Br Heart J 1984; 51: 361.
136. Saumarez RC, Slade AKB, Grace AA, Sadoul N, Camm AJ, McKenna WJ. The significance of paced electrogram fractionation in hypertrophic cardiomyopathy. A prospective study. Circulation 1995, 91: 2762-8.
137. Noble D. The initiation of the heartbeat. Oxford University Press 1979.
138. Goodwin JF, Krikler DM. Arrhythmias as a cause of sudden death in hypertropfic cardiomyopathy. Lancet 1976; 2: 937-40.
139. McKenna WJ, Steward JT, Nihoyannopoulos P, McGinty F, Davies MJ. Hypertrophic cardiomyopathy without hypertrophy: two families with myocardial disarray in the absence of increased myocardial mass. Br Heart J 1990; 63: 287-90.
140. McKenna WJ, Chetty S, Oakley CM, Goodwin JF. Arrhythmias in hypertrophic cardiomyopathy: exercice and 48 hour ambulatory electrocardiographic assessment with and beta adrenergic blocking therapy. Am J Cardiol 1980: 45: 1-5.
141. Kuck KH, Hanrath P, Kunze KP. Programmed electrical stimulation in hypertrophic cardiomyopathy-identification of patients risk for sudden death. Eur Heart J 1983; 4 (Suppl.E): 7.
142. Cecchi F, Olivotto I, Montereggi A, Squillatini G, Dolora A, Maron BJ. Prognosis value of non-sustained ventricular tachycardia and the potential role of amiodarone treatment in hypertrophic cardiomyopathy: assessment in an unselected non-referral based patient population. Heart 1998; 79: 331-6.
143. Kuck KH. Arrhythmias in hypertrophic cardiomyopathy. Pacing Clin Electrophysiol 1997; 20 (Pt.II): 2706-13.
144. Mulrow JP, Healy JR, McKenna MW. Variability of ventricular arrhythmias in hipertrophic cardiomyopathy and implications for treatment. Am J Cardiol 1986; 58: 615-8.
145. Gordon EP, Henderson MA, Rakowski H, Wigle ED. Midventricular obstruction with apical infarction and aneurysm formation. Circulation 1984; 70(Suppl II): II-145.
146. Adabag AS, Casey SA, Kuskowski MA, Zenovich AG, Maron BJ. Spectrum and prognostic significance of arrhythmias on ambulatory Holter electrocardiogram in hypertrophic cardiomyopathy. J Am Coll Cardiol; 2005, 45: 697-704.
147. Morganroth J, Michaelson EL, Horowitz LN, Josephson ME, Pearlman AS. Limitations of routine long-term ECG monitoring to assess ventricular ectopic beats. Circulation 1978; 58: 408-14.
148. Winkle RA. Antiarrhythmic drug effect mimicked by spontaneous variability of ventricular ectopy. Circulation 1978; 57: 1116-21.
149. Michelson EL, Morganroth J. Spontaneous variability of complex ventricular arrhythmias detected by long term ECG recording. Circulation 1980; 61: 690-5.
150. Yoshida N, Ikeda H, Wada T, Matsumoto A, Maki S, Muro A, et al. Exercise-induced abnormal blood pressure responses are related to subendocardial ischemia in hypertrophic cardiomyopathy. J Am Coll Cardiol 1998; 32: 1938-42.
151. Counihan PJ, Frenneaux MP, Webb DJ, McKenna WJ. Abnormal vascular responses to supine exercise in hypertrophic cardiomyopathy.Circulation 1991; 84: 686-96.
152. Sadoul N, Prasad K, Elliot PM, Bannerjee S, Frenneaux MP, McKenna WJ. Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation 1997; 96: 2987-91.
153. Olivotto I, Maron BJ, Montereggi A, Mazzuoli F, Dolara A, Cecchi F. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999; 33: 2044-51.
154. Gilligan DM, Chan WL, Ang EL, Oakley CM. Effects of a meal on hemodynamic function at rest and during exercise in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 1991; 18: 429-36.
155. Fagan TC, Sawyer PR, Gourley LA, Lee JT, Gaffney TE. Postprandial alterations in hemodynamics and blood pressure in normal subjects. Am J Cardiol 1986; 58: 636-41.
156. De Mey C, Hansen-Schmidt S, Enterling D. Postprandial haemodynamic changes: a source of bias in cardiovascular research effected by its own methodological bias. Cardiovasc Res 1988; 22: 703-7.
157. Doi YL, McKenna WJ, Chetty S, Oakley C, Goodwin JF. Prediction of mortality and serious ventricular arrhythmia in hypertrophic cardiomyoapthy. An echocardiographic study. Br Heart J 1980; 44: 150-7.
158. Candell-Riera J, Romero-Farina G, Galve-Basilio E, Palet Balart J, Armadans L, Reina MD, et al. Valor del ecocardiograma Doppler en el pronóstico y en el seguimiento de la miocardiopatía hipertrófica. Rev Esp Cardiol 2001; 54: 7-15.
159. Nistri S, Olivotto I, Betocchi S, Losi MA, Valsecchi G, Pinamonti B, et al. Prognostic significance of left atrial size in patients with hypertrophic cardiomyopathy (from the Italian Registry for Hypertrophic Cardiomyopathy). Am J Cardiol 2006; 98: 960-5.
160. Autore C, Bernabo P, Barilla CS, Bruzzi P, Spirito P. The prognostic importance of left ventricular outflow obstruction in hypertrophic cardiomyopathy varies in relation to the severity of symptoms. J Am Coll Cardiol 2005; 45: 1076-80.
161. McKenna WJ, Iglesias LM. Identificación y tratamiento de los pacientes con miocardiopatía hipertrófica y riesgo de muerte súbita. Rev Esp Cardiol 2000; 53: 123-30.
162. Elliott PM, Gimeno Blanes JR, Mahon NG, Poloniecki JD, McKenna WJ. Relation between severity of left-ventricular hypertrophy and prognosis in patients with hypertrophic cardiomyopathy. Lancet 2001; 357: 420-4.
163. Loui EK, Maron BJ. Hypertrophic cardiomyopathy with extreme increase in left ventricular wall thickness: functional and morphologic features and clinical significance. J Am Coll Cardiol 1986; 8: 57-65.
164. Yamaguchi H, Nakanishi S, Matsumoto S. Hypetrophic cardiomyopathy with giant negative T-clinical and angiographic characteristics. Jap Circ J (Abstracts) 1976; 40: 514.
165. Lewis JF, Maron BJ. Hypertrophic cardiomyopathy characterized by marked hypertrophy of the posterior left ventricular free wall: significance and clinical implications. J Am Coll Cardiol 1991; 18: 421-8.
166. McKenna WJ, Kleinebenne A, Nihoyannopoulos P, Foale R. Echocardiographic measurement of right ventricular wall thickness in hypertrophic cardiomyopathy: relation to clinical and prognostic features. J Am Coll Cardiol 1988; 11: 351-8.
167. Spirito P, Maron BJ, Bonow RO, Epstein SE. Ocurrence and significance of progressive left ventricular wall thinning and relative cavity dilatation in patients with hypertrophic cardiomyopathy. Am J Cardiol 1987; 60: 123-9.
168. Seiler C, Hess OM, Jenni R, Krayenbuelhl HP. Left ventricular chamber dilatation in hypertrophic cardiomyopathy. Prevalence in medically and surgically treated patients. Circulation 1993; 88 (suppl I): I-212.
169. Bingisser R, Candinas R, Schneider J, Hess OM. Risk factors for systolic dysfunction and ventricular dilatation in hypertrophic cardiomyopathy. Int J Cardiol 1994; 44: 225-33.
170. Biagini E, Coccolo F, Ferlito M, Perugini E, Rocchi G, Bacchi-Reggiani L, et al. Dilated-hypokinetic evolution of hypertrophic cardiomyopathy: prevalence, incidence, risk factors, and prognostic implications in pediatric and adult patients. J Am Coll Cardiol 2005; 46: 1543-50.
171. D'Andrea A, Caso P, Severino S, Cuomo S, Capozzi G, Calabro P, et al. Prognostic value of intra-left ventricular electromechanical asynchrony in patients with hypertrophic cardiomyopathy. Eur Heart J 2006; 27: 1311-8.
172. O'gara PT, Bonow RO, Maron BJ, Damske BA, Van Lingen A, Bacharach SL, et al. Myocardial perfusion abnormalities in patients with hypertrophic cardiomyopathy: assessment with thallium-201 emission computed tomography. Circulation 1987; 76: 1214-23.
173. Pitcher D, Wainwright R, Maisey M, Curry P, Lowton E. Assessment of chest pain in hypertrophic cardiomyopathy using exercise thallium-201 myocardial scintigraphy. Br Heart J 1980; 44: 650-6.
174. Uldelson JE, Bonow RO, O'gara PT, Maron BJ, Van Lingen A, Bacharach SL, et al. Verapamil prevents silent myocardial perfusion abnormalities during exercise in asymptomatic patients with hypertrophic cardiomyopathy. Circulation 1989; 79: 1052-60.
175. Cannon RO, Dilsizian V, O'gara PT, Udelson JE, Schenke WH, Quyyumi A, et al. Myocardial metabolic, hemodynamic, and electrocardiographic significance of reversible thallium-201 abnormalities in hypertrophic cardiomyopathy. Circulation 1991; 83: 1660-7.
176. Cannon RO, Dilsizian V, O'gara PT, Udelson JE, Tucker E, Panza JA, et al. Impact of surgical relief of outflow obstruction on thallium perfusion abnormalities in hypertrophic cardiomyopathy. Circulation 1992; 85: 1039-45.
177. Dilsizian V, Smeltzer WR, Dextras R, Simon TR, Bonow RO. Regional thallium abnormalities in hypertrophic cardiomyopathy: myocardial ischemia or distriburbed cellular active cation uptake? Circulation 1990; 82: 9.
178. Lazzeroni E, Picano E, Morozzi L, Maurizio AR, Palma G, Ceriati R, et al. Dipyridamole-induced ischemia as a prognostic marker of future adverse cardiac events in adult patients with hypertrophic cardiomyopathy. Echo Persantine Italian Cooperative (EPIC) Study Group, Subproject Hypertrophic Cardiomyopathy. Circulation 1997; 96: 4268-72.
179. Romero-Farina G, Candell-Riera J, Galve E, Armadans L, Ramos F, Castell J, et al. Do myocardial perfusion SPECT and radionuclide angiography studies in adult patients with hypertrophic cardiomyopathy have prognostic implications? J Nucl Cardiol 2004;11: 578-86.
180. Nagata S, Park YD, Ninanikawa T, Kozuka T, Sakakibara H, Nimura Y. Thallium perfusion and cardiac enzyme abnormalities in patients with familial hypertrophic cardiomyopathy. Am Heart J 1985; 109: 1317-22.
181. Maron BJ, Epstein SE, Roberts WC. Hypertrophic cardiomyopathy and transmural myocardial infarction without significant atherosclerosis of the extramural coronary arteries. Am J Cardiol 1979; 43: 1086-102.
182. Yamada M, Elliot PM, Gane J. Relation of thallium-201 perfusion abnormalities to clinical and prognostic markers in hypertrophic cardiomyopathy. Eur Heart J 1995; 16: 105.
183. Koga Y, Yamaguchi R, Ogata M, Kihara K, Toshima H. Decreased coronary vasodilatory capacity in hypertrophic cardiomyopathy determined by split-dose thallium-dipyridamole myocardial scintigraphy. Am J Cardiol 1990; 65: 1134-9.
184. Romero-Farina G, Candell-Riera J, Péreztol-Valdés O, Castell J, Aguade S, Galve E, et al. SPET miocárdico de perfusión y ventriculografía isotópica en la miocardiopatía hipertrófica obstructiva y no obstructiva. Rev Esp Med Nucl 2001; 20: 530-6.
185. Candell J, Palet J, Castell J, Aguadé S. Exercise thallium-201 scintigraphy in hypertrophic cadiomyopathy. J Nucl Cardiol 1995; 26(Part 2), p19-413: S104.
186. Takeishi Y, Chiba J, Abe Sh, Tonooka I, Komatani A, Tomoike H. Heterogeneous myocardial distribution of iodine-123 15-(p-iodophenyl)-3-R,S- methylpentadecanoic acid (BMIPP) in patients with hypertrophic cardiomyopathy. Eur J Nucl Med 1992; 19: 775-82.
187. Kurata Ch, Tawarahara K, Taguchi T. Myocardial emission computed tomography with iodine-123-labeled beta-methyl-branched fatty acid in patients with hypertrophic cardiomyopathy. J Nucl Med 1992; 33: 6.
188. Kobayashi H, Nakata T, Han S, Takahashi N, Hashimoto A, Tsuchihashi K, et al. Fatty acid metabolic and perfusion abnormalities in hypertrophied myocardium assessed by dual tracer tomography using thallium-201 and iodine-123-beta- methylpentadecanoic acid]. J Cardiol 1994; 24: 35-43.
189. Torizuka K, Yonekura Y, Nishimura T. Phase 3 study of methyl-p-(123I)-iodophenyl pentadecanoic acid, a myocardial imaging agent for evaluating fatty acid metabolism-a multicenter trial. Jpn J Nucl Med 1992; 29: 423.
190. Shimonagata T, Nishimura T, Uehara T, Hayashida K, Kumita S, Ohno A, et al. Discrepancies between myocardial perfusion and free fatty acid metabolism in patients with hypertrophic cardiomyopathy. Nucl Med Commun 1993; 14: 1005-13.
191. Grover-Mckay M, Schwaiger M, Krivokapich J, Perloff F, Phelps ME, Schelbert HR. Regional myocardial blood flow and metabolism at rest in mildly symptomatic patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 1989; 13: 317-24.
192. Geltman EM, Smith JL, Beecher D, Ludbrook PA, Ter-Pogossian MM, Sobel BE. Altered regional myocardial metabolism in congestive cardiomyopathy detected by positron tomography. Am J Med 1983; 74: 773-85.
193. Sochor H, Shelbert HR, Schwaiger M. Studies of fatty acid metabolism with positron emission tomography in patients with cardiomyopathy. Eur J Nucl Med 1986;12: S66-S69.
194. Kurata C, Kobayashi A, Yamazaki N. Dual autoradiographic study with thallium-201 and radioiodinated fatty acid in cardiomyopathy hamsters. J Nucl Med 1989; 30: 80-7.
195. Nakata T, Tanaka Sh, Hamagami Sh, Miyamoto K, Oh-hori K, Iimura. Detection of impaired fatty acid metabolism and dyskinesis in hypertrophic cardiomyopathy with iodine-123-BMIPP. J Nucl Med 1996; 37: 1679-81.
196. Nishimura T, Nagata S, Uehara T, Hayashida K, Mitani I, Kumita ShI. Assessmente of myocardial damage in dilated-phase hypertrophic cardiomyopathy by using indium-111-antimyosin Fab myocardial ascintigraphy. J Nucl Med 1991; 32: 1333-7.
197. Beder SD, Gutgesel HP, Mullins CE, McNamara DG. Progression from hypertrophic obstructive cardiomyopathy to congestive cardiomyopathy in a child. Am Heart J 1982; 104: 155-6.
198. Ten Cate FJ, Roeland J. Progression to left ventricular dilatation in patients with hypertrophic obstructive cardiomyopathy. Am Heart J 1980; 90: 753.
199. Casset-Senon D, Philippe L, Cosnay P, Lopez-Moutault J, Turot-Bracq V, Fauchier JP, et al. Étude isotopique de la perfusion et de línnervation myocardique chez 28 patients atteints de cardiomyopathie hypertrophique promitive: relation avec les troubles du rythme ventriculaire. Arch Mal Coeur 1994; 87: 475-83.
200. Shimizu M, Sugihara N, Kita Y, Shimizu K, Horita Y, Nakajima K, et al. Long-term course and cardiac sympathetic nerve activity in patients with hypertrophic cardiomyopathy. Br Heart J 1992; 67: 155-60.
201. Fujiwara Y, Hamada M, Mukai M. Different myocardial distribution patterns between iodine-123 metaiodobenzylguanidine and thallium-201 in hypertrophic cardiomyopathy. Am J Noninvas Cardiol 1992; 6: 177.
202. Schafers M, Dutka D, Rhodes CG, Lammertsma AA, Hermansen F, Schober O, et al. Myocardial presynaptic and postsynaptic autonomic dysfunction in hypertrophic cardiomyopathy. Circ Res 1998; 82: 57-62.
203. Hiasa G, Hamada M, Saeki H, Ogimoto A, Ohtsuka T, Hara Y, et al. Cardiac sympathetic nerve activity can detect congestive heart failure sensitively in patients with hypertrophic cardiomyopathy. Chest 2004; 126: 679-86.
204. Bonow RO, Frederick TM, Bacharach SL, Green MV, Goose PW, Maron BJ, et al. Atrial systole and left ventricular filling in hypertrophic cardiomyopathy: effect of verapamil. Am J Cardiol 1983; 51: 1386-91.
205. Betocchi S, Bonow RO, Bacharach SL, Rosing DR, Maron BJ, Green MV. Isovolumic relaxation period in hypertrophic cardiomyopathy: assessment by radionuclide angiography. J Am Coll Cardiol 1986; 7: 74-81.
206. Bonow RO, Rosing DR, Bacharach SL, Green MV, Kent KM, Lipson LC, et al. Effects of verapamil on left ventricular systolic function and diastolic filling in patients with hypertrophic cardiomyopathy. Circulation 1981; 64: 787-96.
207. Bonow RO, Dilsizian V, Rosing DR, Maron BJ, Bacharach SL, Grenn MV. Verapamil-induced inprovement in left ventricular diastolic filling and increased exercise tolerance in patients with hypertrophic cardiomyopathy. Short and long term effects. Circulation 1985; 72: 853-64.
208. Candell-Riera J. Papel de las técnicas no invasivas (electrocardiograma, Holter. Prueba en tabla basculante, resonancia magnética nuclear, isótopos) en la evaluación de los pacientes con miocardiopatía hipertrófica. Rev Esp Cardiol 1995; 48: 828-36.
209. Newman H, Sugrue D, Oakley CM, Goodwin JF, McKenna WJ. Relation of left ventricular function and prognosis in hypertrophic cardiomyopathy: an angiographic study. J Am Coll Cardiol 1985; 5: 1064-74.
210. Chikamori T, Dickie Sh, Poloniecki JD, Myers MJ, Lavender JP, McKenna WJ. Prognostic significance of radionuclide-assessed diastolic function in hypertrophic cardiomyopathy. Am J Cardiol 1990; 65: 478-82.
211. O'hanlon R, Assomull RG, Prasad SK. Use of cardiovascular magnetic resonance for diagnosis and management in hypertrophic cardiomyopathy. Curr Cardiol Rep 2007; 9: 51-6.
212. Choudhury L, Mahrholdt H, Wagner A, Choi KM, Elliott MD, Klocke FJ, et al. Myocardial scarring in asymptomatic or mildly symptomatic patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 40: 2156-64.
213. Moon JC, McKenna WJ, McCrohon JA, Elliott PM, Smith GC, Pennell DJ. Toward clinical risk assessment in hypertrophic cardiomyopathy with gadolinium cardiovascular magnetic resonance. J Am Coll Cardiol 2003; 41: 1561-7.
214. Moon JC. ¿Qué significa la captación tardía de gadolinio en la miocardiopatía hipertrófica? Rev Esp Cardiol 2007, 60: 1-4.
215. Maron BJ, Kragel AH, Roberts WC. Sudden death in hypertrophic cardiomyopathy with normal left ventricular mass. Br Heart J 1990; 63: 308-10.
216. Olsen EGJ. Anatomic and light microscopic characterisation of hypertrophic obstructive and non-obstructive cardiomyopathy. Eur Heart J 1983; 4(Suppl F): 1.
217. Maron BJ, Anan TJ, Roberts WC. Quantitative analysis of the distribution of cardiac muscle cell disorganisation in the left ventricular wall of patients with hypertrophic cardiomyopathy. Circulation 1981; 63: 882-94.
218. Maron BJ, Roberts WC. Quantitative analysis of cardiac muscle cell disorganisation in the ventricular septum of patients with hypertrophic cardiomyopathy. Circulation 1979; 59: 689-706.
219. Teare D. Asymmetrical hypertrophy of the heart in young adults. Br Heart J 1958; 20: 1-8.
220. Cohen J, Effat H, Goodwin JF, Oakley CM, Steiner RE. Hypertrophic obstructive cardiomyopathy. Br Heart J 1964; 26: 16-32.
221. Shirani J, Pick R, Roberts W, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol 2000; 35: 36-44.
222. Varnava AM, Elliott PM, Baboonian C, Davison F, Davies MJ, McKenna WJ. Hypertrophic cardiomyopathy: histopathological features of sudden death in cardiac troponin T disease. Circulation 2001; 104: 1380-4.
223. Varnava AM, Elliott PM, Mahon N, Davies MJ, McKenna WJ. Relation between myocyte disarray and outcome in hypertrophic cardiomyopathy. Am J Cardiol 2001; 88: 275-9.
224. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies. Circulation 2006; 113: 1807-16.
225. Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, et al. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell 1990; 62: 999-1006.
226. Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, et al. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell 1994; 77: 701-12.
227. Moolman JC, Corfield VA, Posen B, Ngumbela K, Seidman C, Brink PA, et al. Sudden death due to troponin T mutations. J Am Coll Cardiol 1997; 29: 549-55.
228. Kimura A, Harada H, Park JE, Nishi H, Satoh M, Takahashi M, et al. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet 1997; 16: 379-82.
229. Watkins H, Conner D, Thierfelder L, Jarcho JA, MacRae C, McKenna WJ, et al. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet 1995; 11: 434-7.
230. Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, et al. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet 1996,13: 63-9.
231. Mogensen J, Klausen IC, Perdersen AK, Egeblad H, Bross P, Kruse TA, et al. Alpha-cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J Clin Invest 1999; 103: R39-43.
232. Anan R, Greve G, Thierfelder L, Watkins H, McKenna WJ, Solomon S, et al. Prognostic implications of novel beta cardiac myosin heavy chain gene mutations that cause familial hypertrophic cardiomyopathy. J Clin Invest 1994; 93: 280-5.
233. Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O'Donoghue A, et al. Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med 1995; 332: 1058-64.
234. Nakajima-Taniguchi C, Matsui H, Fujio Y, Nagata S, Kishimoto T, Yamauchi-Takihara K. Novel missense mutation in cardiac troponin T gene found in Japanese patient with hypertrophic cardiomyopathy. J Mol Cell Cardiol 1997; 29: 839-43.
235. Abchee A, Marian AJ. Prognostic significance of beta-myosin heavy chain mutations is reflective of their hypertrophic expressivity in patients with hypertrophic cardiomyopathy. J Investig Med 1997; 45: 191-6.
236. Fujino N, Shimizu M, Ino H, Okeie K, Yamaguchi M, Yasuda T, et al. Cardiac troponin T Arg92Trp mutation and progression from hypertrophic to dilated cardiomyopathy. Clin Cardiol 2001; 24: 397-402.
237. Anan R, Shono H, Kisanuki A, Arima Sh, Nakao SH, Tanaka H. Patients with familial hypertrophic cardiomyopathy caused by a Phe110Ile missense mutation in the cardiac troponin T gene have variable cardiac morphologies and a favorable prognosis. Circulation 1998; 98: 391-7.
238. Moolman JA, Reith S, Uhl K, Bailey S, Gautel M, Jeschke B, et al. A newly created splice donor site in exon 25 of the MyBP-C gene is responsible for inherited hypertrophic cardiomyopathy with incomplete disease penetrance. Circulation 2000; 101: 1396-402.
239. Watkins H. Hypertrophic cardiomyopathy: from molecular and genetic mechanisms to clinical management. Eur Heart J 2001, (Suppl L): L43-L50.
240. Ackerman MJ. Genetic testing for risk stratification in hypertrophic cardiomyopathy and long QT syndrome: fact or fiction? Curr Opin Cardiol. 2005 May;20(3):175-81.
241. Elliott PM, Poloniecki J, Dickie S, Sharma S, Monserrat L, Varnava A, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 2000; 36: 2212.


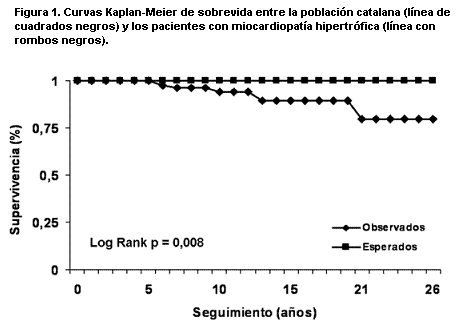{kind=link}
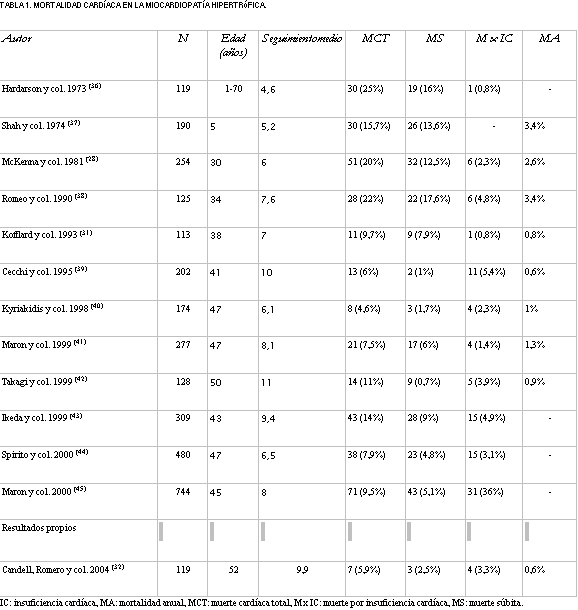{kind=link}
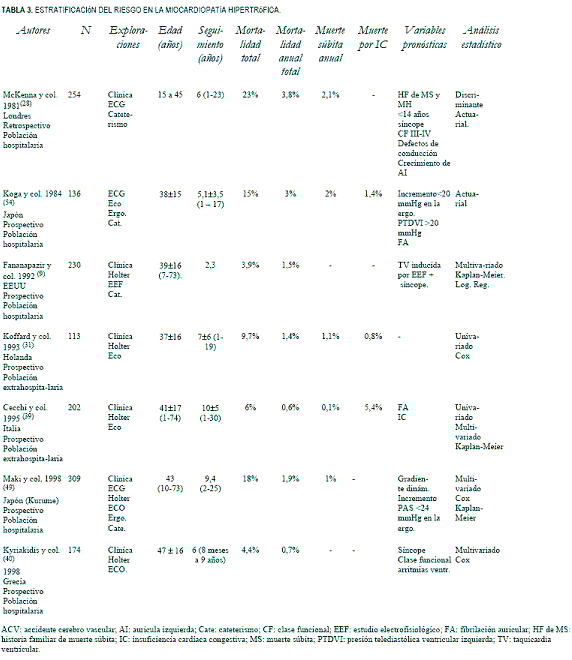{kind=link}
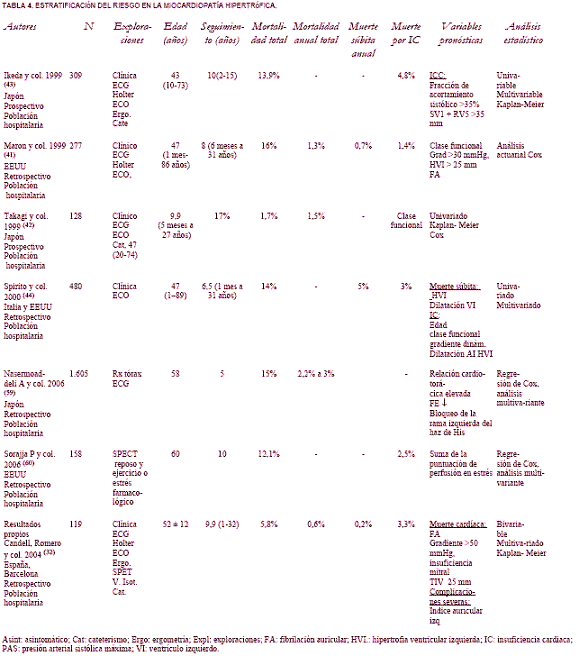{kind=link}
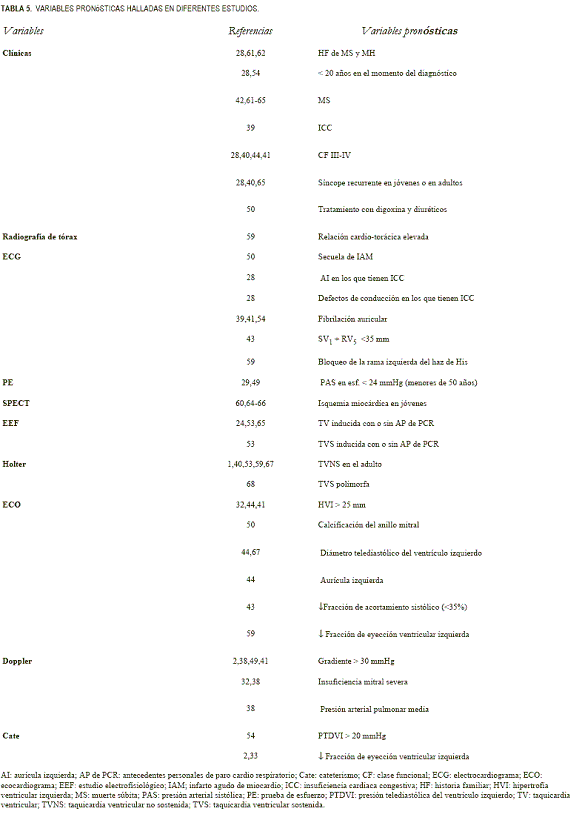{kind=link}
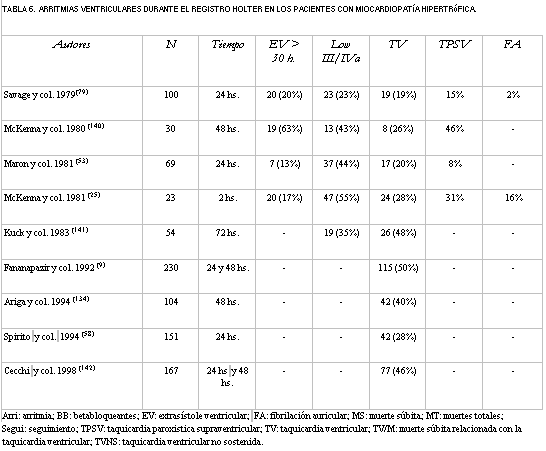{kind=link}