










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkRevista Uruguaya de Cardiología
On-line version ISSN 1688-0420
Rev.Urug.Cardiol. vol.21 no.1 Montevideo Apr. 2006
METODOLOGíA
Cómo analizar un ensayo clínico: el estudio ASCOT-BPLA
DR. EDGARDO SANDOYA 1
1. Centro de Ciencias Biomédicas. Universidad de Montevideo. E-mail: esandoya@um.edu.uy
Un colega comentaba en el ateneo que según el estudio ASCOT-BPLA (1) no se debían usar más los ß-bloqueantes para el tratamiento de la hipertensión arterial, y que a partir de ahora debía indicarse amlodipina en lugar de atenolol. Y se preguntaba "¿cómo estar seguro si eso es así?, ¿ahora debo dejar de usar ß-bloqueantes?"
Es frecuente que en medicina nos enfrentemos a situaciones de este tipo, en que una nueva investigación plantea cambiar una conducta hasta entonces aceptada. ¿Cómo proceder en un caso así? ¿Cómo actuar en este caso concreto?
Para poder tomar cualquier decisión al respecto, en primer lugar debemos conocer en detalle el trabajo que plantea la novedad.
En la actualidad se acepta que la única forma válida de evaluar una terapéutica es mediante un ensayo clínico randomizado apropiado. Los estudios pioneros GISSI (2) e ISIS-2 (3) demostraron el beneficio de los trombolíticos y de la aspirina en el infarto agudo de miocardio (IAM), y comenzó así la era de los grandes ensayos clínicos randomizados. A partir de entonces, diversos ensayos clínicos han dado respuesta a problemas habituales de la cardiología, definiendo el papel de estatinas (4,5), inhibidores ECA (6-8), antiagregantes plaquetarios (9) y de otras terapéuticas (10,11) en su manejo.
El estudio ASCOT-BPLA* fue un ensayo clínico randomizado, pero se necesita saber si su diseño, ejecución y análisis fueron apropiados, para lo que es necesario realizar el análisis crítico del mismo.
Una creencia muy extendida entre los médicos es que para analizar un ensayo clínico se necesita una sólida formación estadística. Ello no es así, pues evaluar un ensayo clínico consiste fundamentalmente en analizar su metodología y esto es algo relativamente simple. Acá se aplica la famosa frase de Einstein: "La mayoría de las ideas fundamentales de la ciencia son esencialmente sencillas y, por regla general, pueden ser expresadas en un lenguaje comprensible para todos".
Actualmente la mayoría de las revistas presentan resúmenes estructurados incluyendo en ellos Introducción, Métodos, Resultados y Comentarios. Mediante su lectura se puede tener idea de los principales aspectos de un ensayo clínico, pero el resumen no permite realizar el análisis crítico, ya que este análisis implica evaluar más información que la que se incluye en el resumen.
El análisis crítico de un ensayo clínico implica considerar cuatro aspectos:
- Saber para qué se hizo el estudio.
- Determinar su validez.
- Analizar sus resultados.
- Ver si es aplicable a la propia realidad
- 1. ¿PARA QUé SE HIZO EL ESTUDIO?
Analizar un ensayo clínico comienza por definir claramente para qué se hizo el mismo, respondiendo para ello a dos preguntas:
- ¿cuál fue el objetivo del estudio?
- ¿cuál fue la variable (endpoint) empleada para evaluar ese objetivo?
1.1 OBJETIVO DEL ESTUDIO
Normalmente un ensayo clínico se centra alrededor de una pregunta, cuanto más clara sea esta, más definitivo va a ser el resultado y su interpretación (12).
En la Introducción de ASCOT-BPLA, se establece "…la aparente falla en la prevención de la cardiopatía isquémica en los ensayos clínicos iniciales se ha atribuido a los diuréticos y ß-bloqueantes empleados. Para una determinada reducción de la presión arterial algunos sugieren que los nuevos antihipertensivos tienen ventaja sobre diuréticos y ß-bloqueantes. Nuestro objetivo fue, por lo tanto, comparar el efecto de combinaciones de atenolol y bendroflumetazida con amlodipina y perindopril sobre el infarto no fatal y la cardiopatía isquémica fatal".
1.2 VARIABLE PRINCIPAL
La variable principal, generalmente única, debe ser capaz de proveer la evidencia clínica más relevante y convincente relacionada al objetivo principal del estudio (13).
La variable principal a analizar en cualquier ensayo clínico cardiovascular debería ser la mortalidad global (12), tal como se hizo en los estudios más importantes de los años 80 y 90. Esto permitió que, entre otras cosas, el debate sobre la hipótesis del colesterol se resolviera cuando el estudio 4S (4) mostró que un hipolipemiante reducía la mortalidad, echando por tierra la duda de que la reducción de mortalidad cardiovascular podría ser anulada por un aumento de la muerte por suicidio y cáncer.
La muerte como variable no requiere ningún análisis especial ya que es objetiva y precisa; la limitante para su empleo es la cantidad de muertes esperadas en el estudio. Cuando la mortalidad es muy elevada, como en la insuficiencia cardíaca clase IV, con pocos pacientes es posible establecer si un tratamiento la reduce, tal como lo hizo el estudio CONSENSUS. En el mismo, en el grupo placebo hubo una mortalidad a seis meses cercana a 50%, por lo que con tan solo en 253 pacientes se pudo demostrar que el enalapril reducía la misma (6). Cuando se estudió su efecto en la insuficiencia cardíaca clase II-III, la que tiene menor mortalidad, se necesitó incluir en el estudio a varios miles de pacientes, y mayor fue el número cuando se estableció el valor de los IECA en la disfunción ventricular izquierda asintomática (8).
En la mayoría de las enfermedades cardiovasculares la mortalidad no es tan elevada, por lo que no es posible el empleo de la mortalidad como variable única. Por ejemplo, si se desea evaluar el efecto de un tratamiento sobre la mortalidad aguda de la angina inestable (situada alrededor de 1%), asumiendo que el mismo pudiera reducirla un 15% en términos relativos (de 1% a 0,85%), el estudio debería incluir al menos a 60.000 pacientes.
Por esa razón los ensayos clínicos utilizan frecuentemente variables combinadas, las cuales incluyen dos o más de ellas, consiguiendo de esa forma una mayor cantidad de eventos con igual número de pacientes.
En ASCOT-BPLA la variable principal fue combinada, incluyendo IAM no fatal y muerte por cardiopatía isquémica.
A diferencia de la muerte, que es una variable fácil de establecer, determinar si se trata de un IAM a veces no es sencillo, existiendo una variación significativa en el número de casos considerados como tales cuando el diagnóstico se define por el investigador o por un comité revisor (14).
Además de la variable principal, el estudio tuvo una serie de variables secundarias que respondieron a sus objetivos secundarios: mortalidad, accidente cerebrovascular (ACV), variable primaria excluyendo IAM silente, muerte cardiovascular, insuficiencia cardíaca, todos los eventos coronarios (angor estable + angor inestable + muerte cardiovascular + IAM + insuficiencia cardíaca) y todos los eventos + procedimientos cardiovasculares (angor estable + angor inestable + muerte cardiovascular + IAM + insuficiencia cardíaca + arritmias potencialmente fatales + arteriopatía periférica + procedimientos de revascularización + trombosis vascular retiniana).
Como vimos, determinar si el evento fue un IAM o no en un ensayo clínico puede resultar complejo. Cuando las variables empleadas son más blandas, como arteriopatía periférica o angina inestable, esa determinación se hace aun más difícil. Las variables que dependen de la decisión del médico tratante, tales como la necesidad de revascularización o la indicación de internación, se asocian a mayor probabilidad de obtener un resultado estadísticamente significativo (15).
Por eso los resultados basados en variables blandas o dependientes de una decisión médica deben ser considerados con mayor cautela. Cuando se analiza un estudio con objetivos combinados que incluyen variables como las blandas o dependientes del médico, es importante ver si la diferencia de eventos no radica exclusivamente en estas sin que exista diferencia en las variables duras, pues en ese caso los resultados serán menos categóricos.
En ASCOT-BPLA se incluyeron variables terciarias, algo muy raro en los ensayos clínicos. Estas comprendieron: IAM silente, angina inestable, angina estable, arteriopatía periférica, arritmias graves, desarrollo de diabetes y desarrollo de falla renal.
2. ¿ES UN ESTUDIO VáLIDO?
Una vez que se tiene claro cuál es el objetivo del estudio y cuál es su variable principal, es necesario determinar si el estudio es válido. Este paso es fundamental pues si el estudio no cumple con los requisitos que determinan su validez, sea cual fuere su resultado, el mismo debe ser descartado.
Para saber si un ensayo clínico es válido es necesario ver si da respuesta a las siguientes preguntas:
- ¿la asignación de los pacientes a cada tratamiento se realizó al azar?;
- ¿fue un estudio doble ciego?;
- ¿al inicio los grupos fueron iguales?;
- ¿se siguió a todos los pacientes y se lo hizo durante un tiempo adecuado?;
- ¿los pacientes se analizaron en el grupo al que fueron randomizados?;
- ¿los dos grupos fueron tratados igual, aparte del tratamiento investigado?
2.1 LA ASIGNACIóN DE LOS PACIENTES A CADA
TRATAMIENTO ¿SE REALIZó AL AZAR?
En Métodos del estudio ASCOT-BPLA, se dice "realizamos un ensayo clínico controlado prospectivo, randomizado, en 19.257 pacientes con hipertensión de 40 a 79 años de edad que tenían al menos otros tres factores de riesgo cardiovascular".
La randomización es esencial para asegurar que la única diferencia entre los dos grupos del estudio consista en el tratamiento que se está investigando. Mediante la distribución al azar se asigna un número de pacientes al grupo que recibe el tratamiento que se investiga (grupo tratado), en este caso amlodipina, y otro número similar al grupo control (en este caso un tratamiento basado en atenolol).
Cuando ningún tratamiento ha demostrado ser efectivo en la enfermedad en estudio, el grupo control recibe placebo; por el contrario, cuando existe un tratamiento efectivo como ocurre en este caso, la comparación se establece con ese tratamiento, pues no sería ético que en ese caso un grupo de pacientes recibiera placebo.
2.2 ¿FUE UN ESTUDIO DOBLE CIEGO?
No se trató de un estudio doble ciego. Se realizó con la metodología PROBE (open treatment and blinded endpoint evaluation), empleada en ensayos clínicos previos con antihipertensivos (16). Esta metodología plantea que aunque la administración de fármacos no sea ciega y no incluya la administración de placebo, el análisis ciego de los resultados, sin saber qué tratamiento recibió el paciente, evita los sesgos.
En este tipo de ensayo clínico los pacientes reciben los medicamentos en su presentación habitual, a diferencia de los ensayos clínicos que emplean placebo, lo que hace que el tratamiento sea más parecido a la práctica clínica habitual.
2.3 ¿AL INICIO LOS GRUPOS FUERON IGUALES?
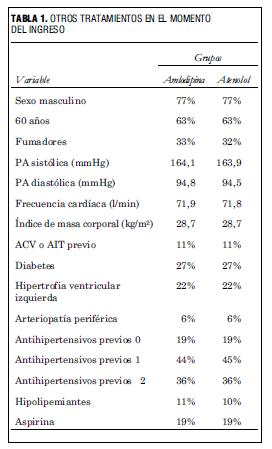
En la tabla 1 se presentan las características de los pacientes en ambos grupos al comienzo del estudio. Como se aprecia, no existió diferencia entre ambos grupos en cuanto a sexo, edad, presión arterial (PA) inicial y demás variables reportadas. Tampoco hubo diferencias en los fármacos que recibían en el momento de ingresar al estudio. Esto habla de que la randomización fue adecuada, pues de no haberlo sido la distribución de las diferentes variables en los dos grupos no hubiera sido similar. Así se consigue que la única diferencia entre los grupos sea el tratamiento en estudio.
2.4 ¿SE SIGUIó A TODOS LOS PACIENTES
Y SE LO HIZO DURANTE UN TIEMPO ADECUADO?
Esta pregunta tiene dos partes. Para encontrar respuesta a la primera parte debemos ver cuántos pacientes fueron incluidos en el estudio y cuántos se presentan en el resultado final. Como puede apreciarse en la figura 1, en el estudio se randomizó a 19.342 pacientes y se excluyó a 85 por irregularidades en la medida de la PA, por lo que quedaron 19.257 evaluables. De ellos, 9.639 fueron asignados al tratamiento basado en amlodipina y 9.618 al tratamiento basado en atenolol.
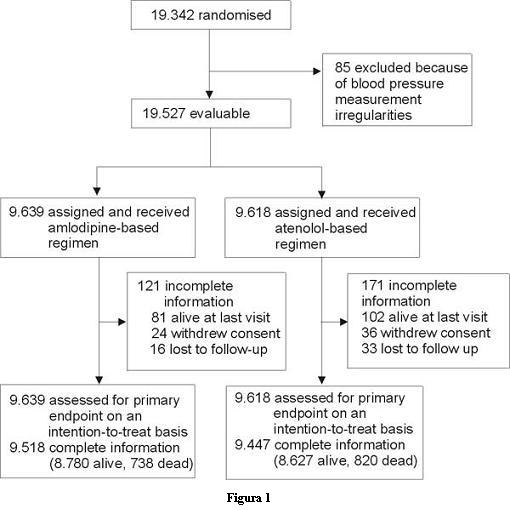
Como sucede en todos los ensayos clínicos, hay pacientes que por diversas razones no completan el mismo; en este caso 24 del grupo amlodipina retiraron el consentimiento y 16 fueron perdidos en el seguimiento, mientras que 36 y 33 casos del grupo atenolol fueron los que no lo completaron por los mismos motivos. Esto hizo que se dispusiera de información completa en 9.518 individuos (99%) del grupo amlodipina y en 9.447 (98%) del grupo atenolol.
En definitiva, si bien no se siguió a todos los pacientes cuando el porcentaje de pérdida es tan pequeño como para afectar el resultado, se considera un seguimiento aceptable.
Ahora debemos buscar respuesta a la segunda parte de la pregunta, que planteaba si se los siguió durante un tiempo adecuado. En Resultados la publicación establece "el estudio fue detenido en forma prematura a una mediana de 5,5 años de seguimiento y acumuló un total de 106.153 pacientes/año de observación" (nota: la variable pacientes/año es la suma de los tiempos de permanencia en el estudio de cada uno de los 19.342 participantes).
El estudio trató de determinar si un tratamiento antihipertensivo era superior a otro en individuos hipertensos con otros tres factores de riesgo, en prevenir el IAM y la muerte coronaria. Cinco años y medio es un plazo adecuado para evaluar la variable principal analizada. Sería inadecuado si en una afección crónica como la hipertensión arterial (HTA) se planteara un seguimiento de pocos meses. En cambio, en un ensayo clínico destinado a evaluar una terapéutica del IAM en su fase aguda, un seguimiento de 30 días es suficiente.
2.5 ¿LOS PACIENTES SE ANALIZARON EN EL GRUPO
AL QUE FUERON RANDOMIZADOS?
Para responder a esta pregunta debemos ver si en el texto se dice que se realizó Análisis por Intención de Tratar de los resultados. Esto significa que a los pacientes asignados al tratamiento basado en amlodipina se los evalúa en ese grupo y a los pacientes asignados a atenolol se los evalúa en ese grupo. En la parte final de Métodos del Resumen encontramos la respuesta "el análisis se realizó por intención de tratar".
Para explicar la lógica de este principio veamos un ejemplo ilustrativo. En un ensayo clínico randomizado que comparó estreptoquinasa con t-PA en el IAM ingresaron simultáneamente al mismo dos pacientes en diferentes centros. El paciente del centro A, asignado a estreptoquinasa, recibió su tratamiento completo. El paciente del centro B, asignado a t-PA, no llegó a recibir el tratamiento pues falleció poco después de incluido en el estudio*, y no se le llegó a administrar el fármaco.
* Un paciente está incluido en el estudio en el momento que es randomizado al grupo tratado o al grupo control.
¿Se debe contar a este último paciente entre los muertos del grupo t-PA? Dado que no recibió el tratamiento, uno se sentiría tentado a decir que no. ¿Diríamos lo mismo si el paciente del grupo estreptoquinasa hubiera fallecido en la mitad de la dosis y no hubiera recibido el total del tratamiento? ¿Y cómo procederíamos si hubiera recibido 90% de la dosis?
Es fácil ver los sesgos que se podrían introducir si el investigador decidiera qué pacientes va a incluir y cuáles no va a incluir en el análisis. Por eso, para que un ensayo clínico sea considerado válido el análisis debe incluir a todos los pacientes que se randomizaron en cada grupo, es decir, debe ser un análisis por intención de tratar, independientemente de que hayan recibido o no el tratamiento.
A uno podría quedarle la duda de si este principio es correcto, dado que se analiza por igual a pacientes tratados y no tratados, pero el mismo se fundamenta en que el azar lleva a que la cantidad de pacientes que reciben tratamiento incompleto en uno y otro grupo sea igual, lo que finalmente equilibra el resultado.
2.6 ¿LOS DOS GRUPOS FUERON TRATADOS IGUAL,
APARTE DEL TRATAMIENTO INVESTIGADO?
No se reporta cuáles fueron los otros tratamientos recibidos durante el estudio además de los que se investigaban. Si bien es importante contar con esta información en estudios con un número tan elevado de pacientes, la adecuada randomización como la que se dio en el estudio que nos ocupa asegura que los otros tratamientos recibidos sean iguales.
Sintetizando las respuestas de las seis preguntas referidas a la validez vimos que:
- la asignación de los pacientes a cada tratamiento se realizó al azar;
- no fue un estudio doble ciego, pero se realizó análisis ciego de las variables;
- al inicio los grupos fueron iguales;
- se siguió a 99% de los pacientes del grupo basado en amlodipina y a 98% del grupo basado en atenolol y se lo hizo durante 5,5 años;
- los resultados se analizaron por el principio de intención de tratar;
- no se reportan los otros tratamientos recibidos durante el estudio.
A partir de estas respuestas podemos concluir que se trata de un estudio válido, pues si bien existe algún aspecto metodológico que no es el ideal, ninguno de ellos introduce sesgos que lleven a invalidar el estudio.
3. ¿CUÁLES SON LOS RESULTADOS?
Una vez que hemos establecido que el estudio es válido, debemos ver cuáles son sus resultados. Es en este punto donde se manejan los dos únicos elementos de estadística que es necesario conocer: el intervalo de confianza (IC) y el valor de p.
Para analizar los resultados del estudio debemos responder a las cuatro preguntas que permiten conocer los beneficios y riesgos del tratamiento investigado:
- ¿cuál es la magnitud del beneficio del tratamiento?;
- ¿cuál es la precisión de la estimación del beneficio?;
- ¿cuál es la magnitud del riesgo del tratamiento?;
- ¿cuál es la precisión de la estimación del riesgo?
3.1 ¿CUáL ES LA MAGNITUD DEL BENEFICIO
DEL TRATAMIENTO?
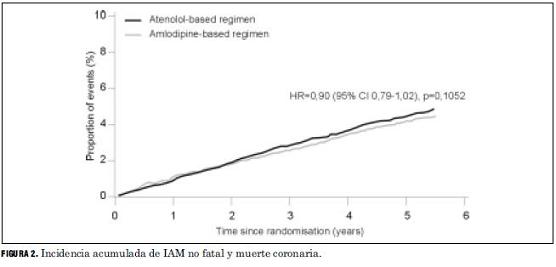
En la figura 2 se aprecian las curvas de incidencia acumulada de IAM no fatal y muerte coronaria para cada tratamiento, y se ve que tuvo un hazard ratio (HR) de 0,90.
El resultado de un ensayo clínico compara la incidencia de la variable en el grupo tratado y la incidencia en el grupo control. Al cociente de la incidencia en el grupo tratado sobre la incidencia en el grupo control se le llama riesgo relativo (RR). A modo de ejemplo, supongamos que la mortalidad con un nuevo tratamiento sea 8% y en el grupo control sea 10%; el RR sería 8 sobre 10, es decir 0,8. Esto significa que el riesgo de muerte con el nuevo tratamiento es 80% con relación al riesgo de muerte en el grupo control.
En ASCOT-BPLA la incidencia de eventos en el grupo amlodipina fue 4,45% (429 eventos en 9.639 pacientes) y en el grupo atenolol 4,92% (474 eventos en 9.618 pacientes). El RR del tratamiento con amlodipina en relación con el atenolol se calcula dividiendo 4,45% sobre 4,92%, o sea que es 0,90. En la publicación el resultado se presenta como HR, que es una forma más sofisticada de expresar el RR. A diferencia del RR calculado en base a los eventos observados al final del estudio, como acabamos de hacerlo, el HR surge del cálculo del RR para cada momento del desarrollo del estudio, pero hacerlo de una u otra manera no arroja diferencia material en el resultado final.
El resultado de los ensayos clínicos puede presentarse de diferentes formas:
- riesgo relativo (RR);
- reducción del riesgo relativo (RRR);
- reducción del riesgo absoluto (RRA);
- número necesario a tratar (NNT).
Estas formas de expresar el resultado surgen de cálculos simples realizados a partir de cuatro números: pacientes en el grupo tratado, eventos en el grupo tratado, pacientes en el grupo control y eventos en el grupo control.
En la tabla 2 se muestra cómo se realizan los cálculos de estas diferentes formas de expresar el resultado. Para interpretar el resultado de un ensayo clínico no es necesario realizar estos cálculos, pero quien lo desee puede ver que los mismos se realizan en forma sencilla:
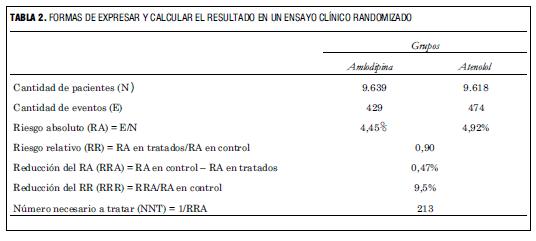
a) el riesgo absoluto (RA) de eventos en cada grupo se calcula dividiendo la cantidad de eventos observados sobre el número de pacientes en ese grupo;
b) el RR, como vimos, compara los riesgos entre los grupos y surge del cociente entre el RA en el grupo tratado y el RA en el grupo control;
c) la RRA expresa cuál es la diferencia absoluta de riesgo entre grupos y se obtiene restando al RA del grupo control el RA del grupo tratado;
d) la RRR expresa cuánto significa la reducción absoluta observada en relación con el RA del grupo control, y se calcula dividiendo la RRA sobre el RA del grupo control;
e) el NNT se calcula dividiendo 1 sobre la RRA (en nuestro caso 1/0,47%).
El NNT indica el número de pacientes que deben ser tratados durante un período similar al que duró el estudio para prevenir un evento (en nuestro caso 5,5 años para prevenir un IAM o una muerte coronaria).
Al analizar el resultado de un estudio se debe prestar especial atención a la forma en que se está expresando dicho resultado, pues se ha demostrado que dependiendo de cómo se lo presente será el impacto que se logre (17). Si en este caso se nos quiere convencer que el tratamiento basado en amlodipina es superior se nos dirá "reduce 10% el infarto o la muerte coronaria". Si, por el contrario, se nos quiere convencer de que no hay diferencias entre tratamientos se nos dirá "la amlodipina no tiene diferencia con el atenolol, reduce el riesgo menos de 1%, apenas 0,47%". En ambos casos se expresan valores que son ciertos, sólo que en el primer caso nos presentan la RRR, que siempre muestra un valor mayor, y en el segundo nos presentan la RRA, la que siempre mostrará un valor más bajo. Por eso es muy importante al ver el resultado de un ensayo clínico saber en qué forma se está expresando el mismo.
Los resultados referidos a los objetivos secundarios y terciarios los analizaremos en el punto 3.2.
3.2 ¿CUáL ES LA PRECISIóN DE LA ESTIMACIóN
DEL BENEFICIO?
Una vez que conocemos cuál es el resultado del tratamiento investigado, debemos analizar con qué precisión se estimó el mismo. Veamos qué significado tienen las dos palabras clave de esta pregunta: precisión y estimación.
Supongamos que queremos saber la talla promedio de los adultos del país. Para poder conocerla deberíamos medir a cada uno de los 2.400.000 de individuos adultos y luego calcular el promedio de las alturas, lo que sería imposible de realizar en la práctica. Una alternativa a ello es seleccionar una muestra representativa de la población adulta y medir su talla. El promedio de altura encontrado en esta muestra nos permitiría estimar la talla media de los adultos de Uruguay. El primer estudio nos permitiría conocer el dato, el segundo nos permitiría estimarlo.
Cuando se realiza una estimación, dependiendo del tamaño de la muestra elegido, habrá una determinada precisión en el dato obtenido. Cuanto mayor sea el tamaño de la muestra, mayor será la precisión del resultado obtenido.
Para conocer cuál es la precisión de la estimación de un resultado se utiliza el IC 95%, el que muestra el rango en el cual estaría el resultado real en 95% de los casos (18). En los estudios con pocos eventos la precisión es menor y el IC 95% es más amplio, mientras que si hay más eventos aumenta la precisión y se estrechan los límites del IC 95%.
Lo que determina la mayor o menor amplitud del IC 95% es la cantidad de eventos y no la cantidad de pacientes del estudio.
ASCOT-BPLA tuvo un HR 0,90 y los límites del IC 95% fueron 0,79 y 1,02, lo que puede verse en forma gráfica en la figura 3. En ella el cuadrado negro representa el valor del HR, y las dos líneas verticales unidas por una horizontal representan los límites del IC 95%. Estos límites caen a uno y otro lado de 1, lo que significa que el resultado real del tratamiento podría ubicarse entre 0,79 (un riesgo de eventos de 79% en los pacientes tratados con amlodipina en relación con el riesgo de los tratados con atenolol) y 1,02 (un riesgo de 102% de eventos de amlodipina en relación con el atenolol). Cuando los límites caen a uno y otro lado de 1 como en este caso, el resultado del estudio no es estadísticamente significativo, pues el efecto real del tratamiento investigado podría ser peor o mejor que el tratamiento del grupo control.
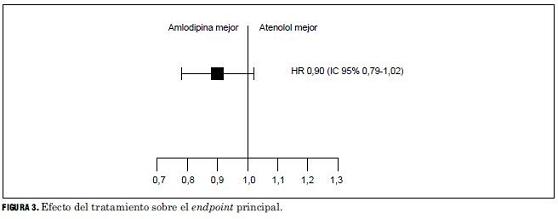
El resultado es estadísticamente significativo cuando los dos límites del IC 95% caen del mismo lado de 1. Cuando ambos están por debajo de 1, el tratamiento investigado es mejor que el tratamiento control, cuando están por encima de 1, el nuevo tratamiento es peor que el tratamiento control.
La significación también puede expresarse como el valor de p, aceptándose que cuando el mismo es menor de 0,05 el resultado es estadísticamente significativo. Es decir que habitualmente en medicina aceptamos como verdad algo que sucede en 95 de cada 100 veces.
En el análisis de los objetivos secundarios y terciarios de ASCOT-BPLA, los investigadores fijaron como estadísticamente significativo un valor de p<0,01 en cambio del 0,05 usual. Hicieron esto para aumentar la certeza de los resultados, pues cuando hay gran cantidad de objetivos, el azar puede hacer que alguno de ellos aparezca como significativo aunque no lo sea (19).
Entre los objetivos secundarios (tabla 3), la mortalidad tuvo una reducción relativa del riesgo de 11%, HR 0,89 (IC 95% 0,81-0,99), p=0,02. Si se toman en cuenta los límites del IC 95%, se ve que este resultado tuvo significación límite debido a la baja prevalencia de ese evento en esta población, lo que lleva a no tener certeza absoluta respecto a este resultado. Esto es refrendado por un valor de p no significativo, pues como vimos para los objetivos secundarios y terciarios se exigía un valor de p<0,01 a los efectos de minimizar el efecto del azar.
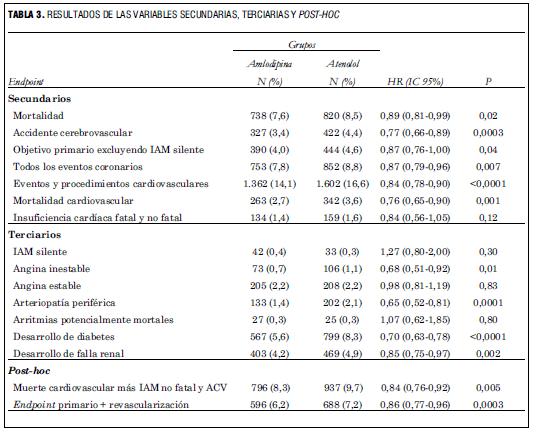
Entre los objetivos secundarios hubo resultados significativos en el ACV (HR 0,77; IC 95% 0,66-0,89; p=0,0003), mortalidad cardiovascular (HR 0,76; IC 95% 0,65-0,90; p=0,001), en todos los eventos coronarios (HR 0,87; IC 95% 0,79-0,96; p<0,007) y en los eventos + procedimientos cardiovasculares (HR 0,84; IC 95% 0,78-0,90; p<0,0001). La reducción observada en la mortalidad cardiovascular deja planteada la duda sobre si pudo haber existido aumento de la mortalidad por alguna otra causa, dado que no hubo descenso significativo de la mortalidad global.
Entre los objetivos terciarios se destaca la reducción en el riesgo de desarrollar diabetes, HT 0,70 (IC 95% 0,60-0,78), con el límite superior alejado de 1 y un valor de p <0,0001, lo que brinda más seguridad acerca de este resultado. También fue significativa la reducción de arteriopatía periférica (HR 0,65; IC 95% 0,52-0,81; p=0,0001) y desarrollo de falla renal (HR 0,85; IC 95% 0,75-0,97; p<0,0001).
En los resultados se introducen dos variables post-hoc: muerte cardiovascular + IAM + ACV y variable primaria + revascularización. Se denomina post-hoc a las variables que no han sido consideradas antes de realizar el estudio y que surgen luego de conocidos los resultados. Los datos proporcionados por este tipo de variables no deben ser tenidos en cuenta, pues cuando se genera una enorme cantidad de información, como la obtenida en este estudio que incluyó casi a 20.000 pacientes, el manejo posterior de la base de datos permite llegar a casi cualquier conclusión que se desee. El único valor de este tipo de variables es el de generar hipótesis, las que luego deberán ser verificadas en un ensayo clínico diseñado para ese fin.
3.3 ¿CUáL ES LA MAGNITUD DEL RIESGO
DEL TRATAMIENTO?
En el estudio no se reporta cuál era el principal efecto secundario a evaluar, pero se presentan datos referidos a la cantidad de pacientes que debieron discontinuar el ensayo por un efecto adverso serio. En base a este número es posible realizar los cálculos en forma similar al cálculo del beneficio, tal como se presenta en la tabla 4.
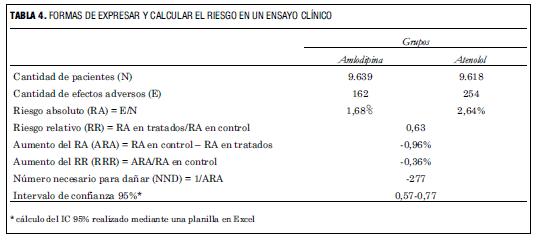
En este caso el aumento del riesgo es negativo (RR 0,63), porque hubo menos eventos adversos (pacientes que abandonaron el estudio por efectos adversos serios) en el grupo basado en amlodipina que en el grupo basado en atenolol.
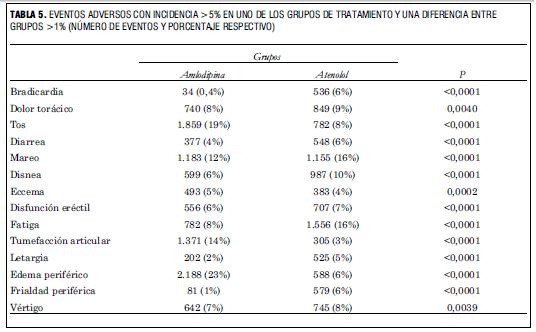
En la tabla 5 se presentan los datos de incidencia de efectos secundarios que se presentaron en más de 5% de los casos y con una diferencia mayor de 1% entre grupos. A partir de los números que allí se presentan es posible calcular el aumento del riesgo de cualquiera de ellos.
3.4 ¿CUáL ES LA PRECISIóN DE LA ESTIMACIóN DEL RIESGO?
Al igual que se hizo con relación al beneficio, se trata de ver cuál es la precisión con que se estimó el riesgo. Mediante una planilla Excel calculamos el IC 95% para el RR 0,63, siendo sus límites 0,57 y 0,77 *. Es decir que la probabilidad de suspender el tratamiento basado en amlodipina fue 63% (entre 57% y 77%) en relación con la probabilidad de suspender el tratamiento basado en atenolol.
* Esta planilla sirve para realizar todos los cálculos acerca de las diferentes formas de expresar el beneficio y el riesgo.
La misma está disponible para quien lo solicite.
4. ¿ES APLICABLE AL CUIDADO DE MIS PACIENTES?
Una vez que sabemos que el estudio es válido y que conocemos cuáles son sus beneficios y sus riesgos, es necesario analizar si el mismo es aplicable a la propia realidad. Para ello se debe responder a las siguientes preguntas:
- ¿el paciente es diferente a los del estudio?;
- ¿el tratamiento es realizable en mi práctica?;
- ¿cuál es el beneficio y los riesgos potenciales para mi paciente?;
- ¿cuáles son los valores y las expectativas del paciente acerca de su enfermedad y su tratamiento?
4.1 ¿EL PACIENTE ES DIFERENTE A LOS DEL ESTUDIO?
Para responder a este interrogante se trata de ver si edad, sexo y características clínicas son similares a las de los pacientes de nuestra propia realidad. Analizando los criterios de inclusión del estudio en la sección Métodos, vemos que debían tener entre 40 y 79 años, que fueran de ambos sexos (finalmente se incluyeron 23% de mujeres y 77% de hombres), tenían hipertensión no tratada con cifras de PA sistólica ³160 mmHg y/o de PA diastólica ³100 mmHg, o hipertensión tratada con uno o más fármacos y PA sistólica ³140 mmHg y/o PA diastólica ³90 mmHg.
Además de la HTA debían tener al menos tres factores de riesgo cardiovascular entre los siguientes (20):
- hipertrofia ventricular izquierda (por electrocardiograma (ECG) o ecocardiograma) en los últimos dos meses;
- alteraciones del ECG: sobrecarga ventricular izquierda, ondas Q anormales, BCRI, cambios de ST-T;
- diabetes tipo 2;
- arteriopatía periférica;
- accidente cerebrovascular incluyendo AIT ³3 meses antes;
- edad ³55 años;
- microalbuminuria/proteinuria;
- tabaquismo;
- relación colesterol total/HDL ³6;
- antecedentes de enfermedad coronaria en familiares de primer grado, antes de los 55 años en hombres y de los 60 años en mujeres.
El otro elemento a tener en cuenta para analizar la similitud o no de los pacientes es conocer cuáles fueron los criterios de exclusión del estudio. Estos comprendieron:
- contraindicación a alguno de los fármacos en estudio;
- HTA maligna;
- HTA secundaria;
- IAM previo o angina en tratamiento;
- ACV, AIT o cirugía cerebral <90 días;
- recibir bloqueantes cálcicos, I-ECA, ß-bloqueantes o diuréticos por alguna razón;
- triglicéridos en ayunas ³4,5 mmol/l;
- BAV de 2º o 3er grado;
- insuficiencia cardíaca clínica (Clase II-IV);
- arritmias no controladas;
- enfermedades importantes hematológicas, gastrointestinales, hepáticas, renales u otra enfermedad que, en opinión del investigador, pudiera interferir con el tratamiento o la capacidad del paciente para completar el estudio;
- alcoholismo, abuso de drogas, psicosis, personalidad antagónica, poca motivación u otro problema emocional o intelectual que pudieran invalidar el consentimiento informado o limitaran su capacidad para cumplir con los requerimientos del protocolo;
- participación en otros estudios de investigación;
- embarazo o lactancia.
De la lectura de los criterios de inclusión y exclusión surge que se trata de pacientes similares a los que podemos ver en nuestro medio.
Algo fundamental a tener en cuenta en la práctica clínica es que los resultados de un ensayo clínico se aplican a pacientes similares a los del estudio. Por lo tanto los resultados de ASCOT-BPLA no son aplicables, por ejemplo, a hipertensos mayores de 79 años, o que no tuvieran al menos otros tres factores de riesgo (como se los define en el estudio) o que tuvieran IAM previo, angina, insuficiencia cardíaca o cualquiera de los criterios de exclusión.
4.2 ¿EL TRATAMIENTO ES REALIZABLE EN SU PRáCTICA?
Esta pregunta apunta a evaluar si se dispone del tratamiento, si existen condiciones generales apropiadas para su empleo y se está entrenado en el mismo.
En el caso que nos ocupa el tratamiento consistía en amlodipina, atenolol, perindopril, doxazosina GITS, bendroflumetazida y potasio. Si bien en nuestro medio no se dispone de bendroflumetazida, este diurético es una tiazida que es comparable a la hidroclorotiazida. Por otro lado tampoco existe la doxazocina GITS, pero cuando uno analiza la cantidad de pacientes tratados con cada fármaco en el estudio no se menciona que hubiera pacientes que recibieran este fármaco (tabla 6), el que, por otro lado, se hallaba recién en el quinto escalón de tratamiento (figura 4).
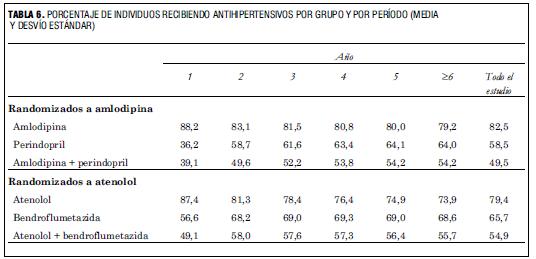
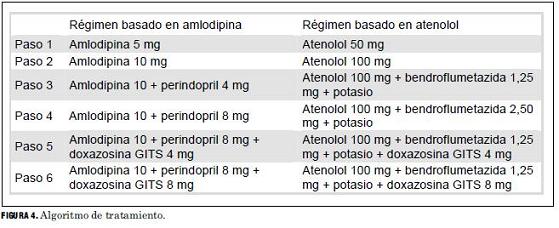
Por lo tanto se trata de un tratamiento disponible en nuestro medio y que no requiere condiciones especiales ni entrenamiento específico para su realización. Este punto puede ser muy importante en un ensayo clínico que propone un tratamiento basado en tecnología sofisticada o que requiera de equipamiento no disponible en el medio, o de un operador con entrenamiento que sólo se consigue en centros de excelencia a nivel mundial.
4.3 ¿CUáL ES EL BENEFICIO Y LOS RIESGOS POTENCIALES PARA MI PACIENTE?
Esta pregunta no tiene sentido cuando se está realizando el análisis del estudio, pero es muy importante en el momento de decidir si administramos el tratamiento a un paciente.
Se trata de ver si este paciente es de los más sanos del estudio (40 años, con menos riesgo cardiovascular global) o de los más enfermos entre los del estudio (más viejo, con mayor riesgo cardiovascular). En ASCOT-BPLA esta pregunta no es muy relevante, como sí lo es cuando se trata de un tratamiento que puede brindar un importante beneficio pero que tiene probabilidad de efectos secundarios severos. En ese caso, si nuestro paciente no está muy enfermo, tal vez sea mejor no arriesgar un potencial beneficio ante la posibilidad de una complicación seria; por el contrario, si está entre los más enfermos, el nuevo tratamiento puede ser una muy buena opción a pesar de los riesgos que conlleva.
4.4 ¿CUáLES SON LOS VALORES Y LAS EXPECTATIVAS DEL PACIENTE ACERCA DE SU ENFERMEDAD
Y SU TRATAMIENTO?
Esta última pregunta permite que la toma de decisiones incluya todas las variables necesarias, ya que afortunadamente hay una tendencia cada vez mayor a dar participación activa al paciente en la decisión acerca del tratamiento.
Es parte del arte médico distinguir entre quienes desean participar de la decisión y quienes esperan que le indiquemos lo que debe hacer sin más trámite.
Por último, es muy importante la valoración que el paciente hace de su enfermedad, de los sacrificios que está dispuesto a realizar y de los riesgos que acepta correr. Existen pacientes que dicen que están dispuestos a lo que sea con tal de mejorarse, mientras que otros no quieren sacrificar aspectos que entienden importantes para su calidad de vida al hacer determinado tratamiento, y este elemento debe ser muy tenido en cuenta en el momento de decidir.
CONCLUSIONES
Hemos visto que el estudio ASCOT-BPLA fue un ensayo clínico randomizado realizado en hipertensos de 40 a 79 años que tenían tres o más factores de riesgo cardiovascular asociados. El mismo evaluó si un tratamiento basado en amlodipina y perindopril era superior a uno basado en atenolol y bendroflumetazida en relación con la incidencia de IAM no fatal y muerte por cardiopatía isquémica. El tratamiento basado en amlodipina tuvo 10% de reducción de riesgo relativo de la variable principal, pero ello no fue estadísticamente significativo (IC 95% 0,79-1,02), p=0,10. Se observó menor incidencia de ACV (HR 0,77; IC 95% 0,66-0,89; p=0,0003) y menor desarrollo de diabetes (HT 0,70; IC 95% 0,60-0,78; p<0,0001) con el tratamiento basado en amlodipina. Existió menor incidencia de eventos adversos serios que llevaran a discontinuar el tratamiento entre los pacientes que recibieron tratamiento basado en amlodipina que en los tratados en base a atenolol, RR 0,63 (IC 95% 0,57-0,77).
Luego de este análisis estamos en condiciones de decir a nuestro colega que el estudio indicaría que amlodipina (o una combinación de amlodipina y perindopril) es superior a atenolol (o una combinación de atenolol y bendroflumetazida) en la prevención de ACV y del desarrollo de diabetes en hipertensos con alto riesgo cardiovascular, sin cardiopatía isquémica, falla cardíaca ni arritmias graves.
Pero en medicina no deberíamos cambiar nuestra práctica en base al resultado de un solo ensayo clínico. Para asegurarnos que hemos llegado a la verdad acerca de un tratamiento para una determinada enfermedad, idealmente deberíamos disponer de varios ensayos clínicos de diseño, ejecución y análisis de resultados adecuados, realizados en diferentes centros y en diferentes países, y que muestren resultados concordantes.
Mientras esto no suceda debemos integrar la nueva información al cuerpo de evidencia existente respecto a la entidad que nos ocupa, sabiendo que muchas situaciones de la práctica clínica habitual aún no tienen una respuesta definitiva derivada de este tipo de estudios.
BIBLIOGRAFíA
1. Dahlof B, Sever PS, Poulter NR, Wedel H, Beevers DG, Caulfield M, et al; ASCOT Investigators. Prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA): a multicentre randomised controlled trial. Lancet 2005; 366: 895-906.
2. Gruppo Italiano per lo Studio della Streptochinasi nell’Infarto Miocardico. Effectiveness of intravenous thrombolytic treatment in acute myocardial infarction. Lancet 1986; i: 397-402.
3. The ISIS-2 collaborative group. Randomized trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; ii: 349-60.
4. Scandinavian Simvastatin Survival Group. Randomised trial of cholesterol lowering in 4.444 patients with coronary artery disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344: 1383-9.
5. Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N Engl J Med 1995; 333: 1301-7.
6. The CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N Engl J Med 1985; 316: 1429-35.
7. The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med 1991; 325: 293-302.
8. The SOLVD Investigators. Effect of enalapril on mortality and the development of heart failure in patients asymptomatic patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med 1992; 327: 685-91.
9. Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324: 71-86.
10. Yusuf S, Zucker D, Peduzzi P, Fisher LD, Takaro T, Kennedy JW, et al. Effect of coronary artery bypass graft surgery on survival: overview of 10-year results from randomised trials by the Coronary Artery Bypass Graft Surgery Trialists Collaboration. Lancet 1994; 344: 563-70.
11. Hambretch R, Walter C, Mobius-Winkler S, Gielen S, Linke A, Conradi K, et al. Percutaneous coronary angioplasty compared with exercise training in patients with stable coronary artery disease. Circulation 2004; 109: 1371-8.
12. Topol EJ, Califf RM, Van-de-Werf F, Simoons M, Hampton J, Lee KL, et al. Perspectives on large-scale cardiovascular clinical trials for the new millennium: The Virtual Coordinating Center for Global Collaborative Cardiovascular Research (VIGOUR) Group. Circulation 1997; 95(4): 1072-82.
13. Statistical principles for clinical trials. ICH Harmonised Tripartite Guideline. ww.ich.org/LOB/media/MEDIA485.pdf
14. Naslund U, Grip L, Fischer-Hansen J, Gundersen T, Lehto S, Wallentin L. The impact of an end-point committee in a large multicentre, randomized, placebo-controlled clinical trial: results with and without the end-point committee’s final decision on end-points. Eur Heart J 1999; 20(10): 771-7.
15. Freemantle N, Calvert M, Wood J, Eastaugh J, Griffin C. Composite outcomes in randomized trials: greater precision but with greater uncertainty?. JAMA 2003; 289: 2554-9.
16. Hansson L, Lindholm LH, Niskanen L, Lanke J, Hedner T, Niklason A, et al. Effect of angiotensin-converting-enzime inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPPP) randomised trial. Lancet 1999; 353: 611-6.
17. Bucher HC, Weinbacher M, Gyr K. Influence of method of reporting study results on decision of physicians to prescribe drugs to lower cholesterol concentration. BMJ. 1994; 309: 761-4.
18. Davies H. What are confidence interval? www.evidence-based-medicine.co.uk
19. Statistical Analysis Plan, Ascot Study Protocol. www.ascotstudy.org
20. Working Protocol. Ascot Study Protocol. www.ascotstudy.org

