











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
PermalinkSr. Director de la Revista Médica del Uruguay
Prof. Agdo. Dr. Álvaro Danza
Presente
De nuestra mayor consideración
Las coagulopatías congénitas más frecuentes son la hemofilia A (déficit del factor VIII de la coagulación), la hemofilia B (déficit del factor IX de la coagulación) y la enfermedad de von Willebrand (EVW). La hemofilia A (HA) y la hemofilia B (HB) son enfermedades recesivas ligadas al cromosoma X que afectan a 1 en 5.000 y 1 en 30.000 varones, respectivamente, producidas por mutaciones en los genes F8 y F9. Las mujeres pueden ser portadoras asintomáticas o pueden presentar síntomas leves o moderados. Se estima que la hemofilia se hereda en un 70% de los casos, mientras que en otro 30% puede ser originada por mutaciones de novo1.
Se han descrito diferentes tipos de mutaciones en los genes F8 y F9, con más de 3.000 en el F8. Las mutaciones puntuales son las más frecuentes, el 95% de las delecciones se asocian con el fenotipo severo, las inserciones también conducen a hemofilia severa. Las duplicaciones son inusuales. Es posible encontrar también mutaciones que afectan el corte y empalme y mutaciones en la región promotora. En el mundo cerca del 50% de los pacientes con HA severa tienen la inversión del intrón 22 o del intrón 12.
La EVW muestra un patrón de herencia autosómico dominante o recesivo, sin predilección por el sexo; sin embargo, las mujeres pueden presentar síntomas más frecuentemente, producto de la hipermenorrea. En ella se altera cualitativa o cuantitativamente el factor de von Willebrand (FVW) y afecta aproximadamente al 1% de la población general. La Sociedad Internacional sobre Trombosis y Hemostasia la clasifica ya sea por la alteración cuantitativa (tipo 1 y tipo 3) o cualitativa (tipo 2)3.
El tipo 1 es la forma más común de la EVW y representa cerca del 80% de los casos, es transmitido como rasgo autosómico dominante con penetración incompleta. En el tipo 2 se reconocen cuatro distintas formas cualitativas: 2A, 2B, 2M y 2N. La herencia es autosómica dominante en la mayoría de los casos y como rasgo autosómico recesivo en el tipo 2N. La EVW tipo 3 se hereda como rasgo autosómico recesivo. Aproximadamente el 65% de los casos índice con EVW tipo 1 tienen mutaciones en el gen (FVW), de las cuales aproximadamente el 70% son mutaciones missense. Para la enfermedad tipo 2A se han reportado principalmente mutaciones missense que afectan las subunidades D1/D2/D' D3/A2 y CTCK. Con respecto al tipo 2B, se han reportado mutaciones missense en el dominio A1, y variantes que potencian la unión a la glicoproteína a GPIbα. En cuanto al tipo 2M, se han observado mutaciones missense en los dominios A1 y A3, las cuales causan la reducción de la unión a GPIbα (dominio A1) y/o al colágeno (dominio A3). El tipo 2N es causado por mutaciones missense en la secuencia que codifica los dominios D' D3, afectando la unión con el factor VIII. Para el tipo 3, se han encontrado mutaciones en 85% a 90 % de los casos índice, se han descrito delecciones, mutaciones nosense, mutaciones de splicing y missense4.
En Uruguay, desde el año 2020, se comenzó a estudiar la distribución de mutaciones de las principales coagulopatías hereditarias, comenzando con los pacientes con hemofilia A severa de todo el país, donde se ha estimado la frecuencia de las inversiones de los intrones 1 y 22(5). En la región noreste del país ya se han detectado otros tipos de mutaciones en pacientes con hemofilia A; sin embargo, aún falta analizar a la mayoría de las familias para determinar las mutaciones causales en los casos severos, leves y moderados de HA y HB, así como los diferentes tipos de EVW. El GENHEU (Genética en Hemofilia en Uruguay) es el primer análisis genético a nivel nacional de las principales coagulopatías congénitas. Los principales objetivos de esta carta son presentar los resultados alcanzados hasta ahora y dar a conocer este estudio.
La detección de las mutaciones causales de los principales trastornos congénitos de la coagulación sanguínea permite un seguimiento adecuado de los pacientes y sus familias, ya que posibilita la detección del estado de portadoras y el diagnóstico prenatal, ofrece una aproximación a la probabilidad del desarrollo de inhibidores y permite distinguir la hemofilia A de la EVW, así como conocer la distribución de las mutaciones en la población.
Bajo consentimiento informado y contando con la aprobación del Comité de Ética de la Facultad de Humanidades y Ciencias de la Educación de la Udelar, esta investigación busca incluir a todos los pacientes de Uruguay con las principales coagulopatías hereditarias. La extracción del ADN se realiza a partir de muestras de sangre (método salino), la técnica Inverse Shifting PCR, se realiza a los pacientes con hemofilia A severa para descartar las inversiones de los intrones 1 y 22 del gen F8. Esta etapa es realizada en el PDU de Diversidad Genética Humana del Centro Universitario de Tacuarembó-Udelar. A continuación, la detección de otras mutaciones en los casos índice se realiza con el panel de genes MHCD (secuenciación masiva paralela) en la Plataforma Genómica de la Facultad de Ciencias-Udelar, lo que permite secuenciar las regiones codificantes de los genes F8, F9 y FVW con una profundidad promedio de 500X. Posteriormente, las mutaciones patogénicas en los pacientes y en las posibles portadoras son confirmadas por secuenciación Sanger.
Hasta el momento se han analizado 35 pacientes con HA, así como portadoras, con una edad promedio de 32 años (rango de 3 a 72 años) y pertenecientes a 23 familias de seis departamentos del país.
A continuación se muestra la (tabla 1) con los resultados preliminares del estudio GENHEU.
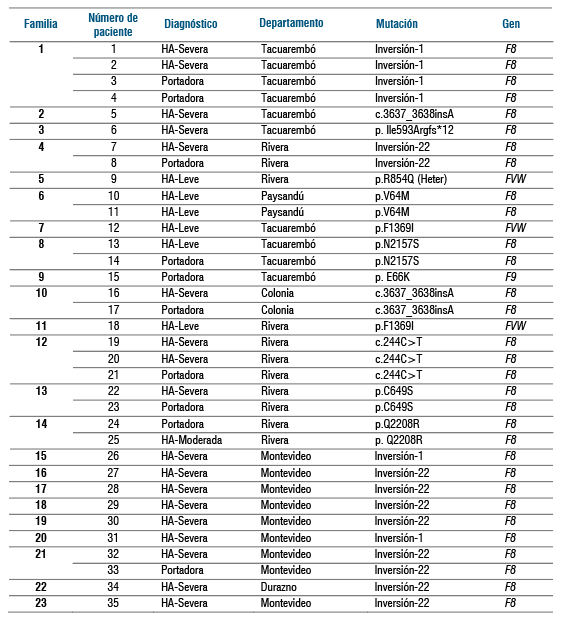
Aparte de la descripción del espectro de mutaciones causales de los principales trastornos de la coagulación congénitos en este grupo de familias de Uruguay, es importante resaltar que tres personas (números 9, 12 y 18) diagnosticadas con HA leve, no mostraron mutaciones en el gen F8, pero sí fueron observadas variantes patogénicas en el gen FVW, por lo que este estudio permitió la reclasificación del diagnóstico. De ellas, la número 9 es portadora de la mutación más frecuente en Europa de EVW tipo 2N (p.R854Q). Esto tiene implicancias a nivel personal para el paciente y a nivel de su seguimiento y tratamiento, así como en el asesoramiento genético para la familia.
Estos datos preliminares permiten caracterizar la etiología genética de la hemofilia y otras coagulopatías congénitas y empezar a conocer la distribución geográfica de las mutaciones causales en Uruguay. Pretendemos seguir estudiando a un mayor número de pacientes, así como realizar el estudio de otras coagulopatías congénitas.
Si algún colega considera necesario el estudio de sus pacientes, puede ponerse en contacto con los investigadores, ya que es de nuestro interés incluir a la mayor cantidad posible.
Sin otro particular, saludan a Ud. atentamente,

