











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Introducción
La displasia septo-óptica (SOD), también conocida como síndrome De Morsier, es un desorden congénito raro, con una incidencia de 2-3/100.000 nacidos vivos1, caracterizada por una combinación de hipoplasia del nervio óptico, disfunción hipofisaria y anormalidades de la línea media. La hipoplasia del nervio óptico puede ser unilateral o bilateral, lo que resulta en grados de visión variables2,3. El hipopituitarismo se observa en 62% a 80%4 de los casos. Según algunos reportes, la deficiencia de hormona de crecimiento es la más frecuente5. El hipogonadismo hipogonadotrófico es un síndrome caracterizado por falla gonadal y niveles de gonadotrofinas bajos. La infertilidad que presentan estos pacientes se debe a una alteración de la secreción de gonadotrofinas, lo que lleva a una falta de estimulación ovárica con anovulación crónica. El diagnóstico temprano del hipogonadismo puede prevenir los efectos negativos sobre esqueleto y metabolismo6. Presentamos un caso clínico de síndrome de De Morsier (displasia septo-óptica) con hipogonadismo hipogonadotrófico, déficit de hormona de crecimiento (GH) y compromiso de la densidad mineral ósea con deseo gestacional.
Caso clínico
Paciente de sexo femenino, 37 años, derivada desde la policlínica de fertilidad. A los 11 años, en el contexto de evaluación por talla baja (debajo de 2 desvíos estándar (DE) de la media para edad y sexo), con hipocrecimiento (velocidad de crecimiento menor a -1 DE por más de un año, acompañado de talla baja) y retraso del desarrollo puberal, se diagnosticó hipogonadismo hipogonadotrófico y déficit de GH. Como etiología, presenta síndrome de De Morsier diagnosticado por resonancia nuclear magnética (RNM) a la misma edad que evidenció ausencia de septum pellucidum, hipoplasia del nervio óptico con hipoplasia de cuerpo calloso, tallo hipofisario y quiasma óptico traccionados ocupando la silla turca. Además, no se reconoce la neurohipófisis (figuras 1,2 y3).
Como antecedentes de la enfermedad actual, presenta dos intervenciones quirúrgicas por labio leporino. Desprendimiento de retina congénito en ojo derecho. Glaucoma y cataratas. Nistagmo ocular bilateral. No presenta retraso a nivel del desarrollo cognitivo, logrando realizar primaria completa a nivel formal educativo.
Exploración complementaria
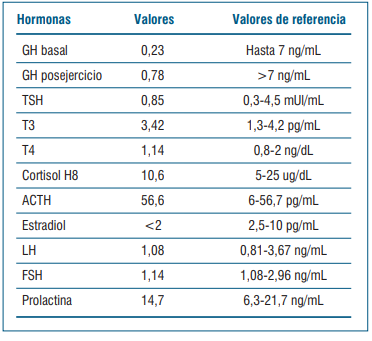
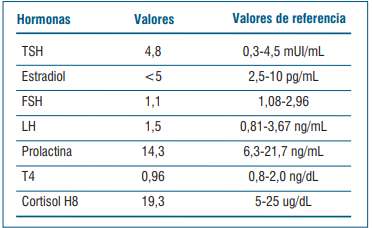
Se realiza dosificación de hormonas evidenciando una respuesta insuficiente de la GH al ejercicio y gonadotrofinas en valores prepuberales (tabla 1). El cortisol basal está en zona gris (entre 3-15 mcg/dl). Se solicitó test de estímulo con hormona adrenocorticotrópica (ACTH) para valorar insuficiencia suprarrenal (cortisol posestímulo de ACTH <18 mcg/dl), que no se realizó. En la evolución se revaloró cortisol y fue suficiente (≥15 mcg/dl).
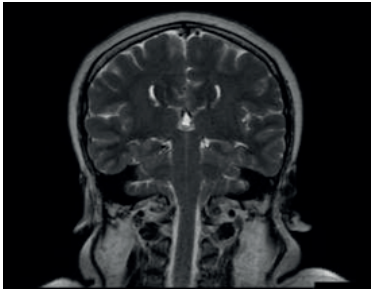
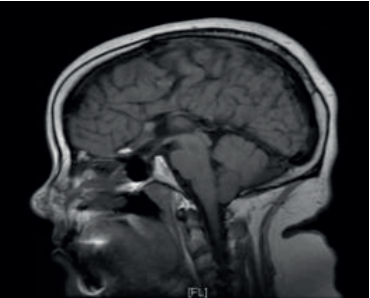
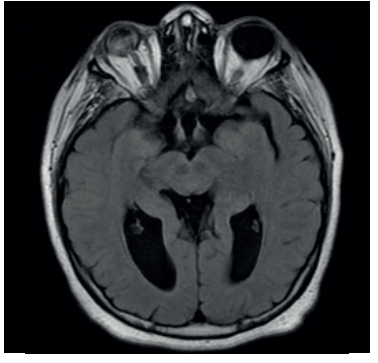
El diagnóstico de déficit de GH se realiza por hipocrecimiento, edad ósea retrasada acorde con 8 años y con test estímulo de GH al ejercicio insuficiente. Dado que presenta alteraciones en la neuroimagen, no fueron necesarios otros test de estímulo para el diagnóstico.
Recibió tratamiento con GH a una dosis de 0,035mg/kg/día desde los 11 hasta los 18 años con muy buena respuesta, alcanzando una talla adulta final de 155 cm.
Se realiza inducción de la pubertad con estradiol por hipogonadismo hipogonadotrófico e impuberismo (ausencia de caracteres sexuales secundarios) y a los 15 años se inició estroprogestágenos.
Recibió acetato de criproterona y etinilestradiol 0,035 mg, que suspende por indicación ginecológica hace un año debido a deseo gestacional, desde entonces se encuentra en amenorrea. Actualmente, desea iniciar tratamiento de fertilidad.
Exploración complementaria actual
Ecografía ginecológica que evidencia un útero bien desarrollado, medidas 51 x 21 x 53 mm con línea endometrial de 2,5 mm regular y homogénea. Ovario derecho 14 x 11 mm e izquierdo 17 x 12 mm, aspecto normal.
No presenta clínica de déficit de hormona antidiurética, presenta ionograma y densidad urinaria sin alteraciones. No alteración de otros ejes hipofisarios (tabla 2).
Presenta una absorciometría de rayos X de energía dual (DXA), con Z score menor a 2 en columna lumbar y cadera. No presentó fracturas patológicas.
Dislipemia mixta en tratamiento nutricional, no realiza actividad física. No presenta otras alteraciones metabólicas.
Al examen físico se observa obesidad grado II de larga data, peso actual 91 kg, talla 155 cm, con un índice de masa corporal (IMC) de 38 kg/m2. A nivel de piel se destaca acantosis nigricans en pliegues.
Como alteraciones de línea media presenta labio leporino, ojo derecho de tamaño menor que el contralateral, nistagmo ocular bilateral, paladar ojival sin otras dismorfias ni alteraciones neurológicas. Mamas y vello genital: Tanner V.
Discusión
La displasia SOD es una entidad heterogénea con manifestaciones clínicas y radiológicas que varían en un extenso rango, por lo que se plantea que se trata de un síndrome. Agentes etiológicos planteados incluyen infecciones virales, insultos vasculares, edad materna y mutaciones genéticas. La mayoría de los casos de SOD son esporádicos, pero casos familiares han sido descriptos asociados a mutaciones en el desarrollo de genes: HESX1, SOX2, SOX3, OXT2, PROKR2, FGF1 y FGF8. Estas mutaciones son identificadas en menos del 10% de los casos, lo que sugiere que la etiología sea multifactorial con la participación de otros genes, factores ambientales y epigenéticos1.
La clínica es muy variada. Pueden ocurrir alteraciones visuales, neurológicas y endócrinas. Las alteraciones visuales ocurren secundariamente a la hipoplasia del nervio óptico, que van desde un daño parcial en la visión hasta ceguera, también se asocia a estrabismo y nistagmo. Esta paciente presentó desprendimiento de retina congénito en ojo derecho y nistagmo ocular bilateral. Las secuelas neurológicas comprenden retraso en el desarrollo cognitivo, convulsiones, hasta incluso parálisis cerebral. Cuando se tiene al menos una sospecha de SOD, que en esta paciente se evidenció por el fenotipo al nacimiento, labio leporino, trastorno ocular y nistagmo, se debe solicitar una RNM para evaluar anormalidades del hipotálamo-hipófisis y posibles defectos de la línea media5. Para el diagnóstico, deben estar presentes dos de tres criterios: ausencia del septum pellucidum, disgenesia del cuerpo calloso e hipoplasia del nervio óptico. Otros hallazgos posibles son microftalmia o anoftalmia, coloboma, hipófisis ectópica o hipoplasia, tallo hipofisario hipoplásico7. Los ejes hipofisarios más frecuentemente afectados son el somatotropo, corticotropo, tirotropo y déficit de hormona antidiurética (ADH). Esta paciente se presentó con un déficit de GH y un hipogonadismo hipogonadotrófico. En un estudio realizado en Estados Unidos, en 1984, observaron que de una muestra de 16 pacientes con SOD en un seguimiento de 3,5 años. El orden y grado de afectación de los ejes hormonales era similar a lo reportado en la literatura hasta el momento. Encontraron que 73% presentaba déficit GH aislada, y 34% de deficiencia de ACTH. Le siguen el déficit de hormona tirotropa (TSH) y ADH en 24% y 21%, respectivamente8. Se concluye que los déficits hormonales son frecuentes y pueden progresar en el tiempo, por lo que la evaluación endocrinológica regular es necesaria para la correcta sustitución y prevención de comorbilidades asociadas al hipopituitarismo.
La mayoría de las neuronas secretoras de la hormona liberadora de gonadotropina (GnRH) se encuentran en el área preóptica del hipotálamo, migran a este lugar en la primera semana de gestación. Una alteración estructural adyacente o en dicha área, previa a la semana, podría generar una diminución de dichas neuronas o una alteración en su funcionamiento.
La injuria por SOD en la línea media ocurre entre las semanas 5 y 8 de gestación1,9. En este caso, se podría pensar que esta fue una posible causa, ya que la paciente presenta múltiples alteraciones a dicho nivel e hipogonadismo hipogonadotrófico.
En niñas no existe clínica para poder sospechar déficit de GnRH. Sin embargo, en niños se puede inferir mediante el crecimiento del pene y testículos, lo que nos podría orientar rápidamente al diagnóstico. Existe un período, llamado minipubertad, que ofrece una oportunidad temprana para el diagnóstico en ambos sexos6.
Las pacientes que presentan HH tienen niveles bajos de FSH, LH, hormona antimulleriana (AMH) y volumen ovárico que están asociados a la disminución de la cantidad de folículos antrales y a su maduración, lo que resulta en anovulación crónica6.
La inducción de la ovulación en estas pacientes, mediante la administración de FSH recombinante, lleva a un buen resultado en la fertilidad10 en ausencia de otros factores, como edad mayor a 35 años o alteración de la infertilidad en el factor masculino. Ésta puede ser lograda con la administración pulsátil de la GnRH o directamente con gonadotrofinas. El tratamiento con GnRH pulsátil restaura la secreción de gonadotrofinas de manera fisiológica y su mayor ventaja, comparada con la administración de gonadotrofinas, es que disminuye el riesgo de embarazo múltiple y evita la hiperestimulación del ovario. Esta terapia debe ser considerada de primera línea en pacientes con HH congénito (siempre que las células gonodotrópicas hipofisarias estén indemnes).
El resultado de la inducción de la ovulación en estas pacientes es excelente, alcanzando 90% de ovulación por ciclo y 27,6% de concepción por ciclo de ovulación. La fertilización in vitro es una alternativa si la concepción falla luego de varios ciclos de ovulación exitosos6. En Uruguay existe una ley que reglamenta la reproducción humana asistida y que está incluida en el Plan Integral de Atención en Salud (PIAS)11. El PIAS consiste en prestaciones que están bajo la cobertura universal del Sistema Nacional Integrado de Salud.
Sin embargo, para una fertilidad óptima es necesario un correcto desarrollo uterino, que en esta paciente fue así. Cuando se realiza el diagnóstico de retraso puberal (ausencia de talarquia a los 13 años de edad o ausencia de menarca luego de tres años de la aparición del botón mamario), como en el caso de esta paciente, se debe realizar la inducción de la misma para emular la fisiología. Esto se logra con la administración de estrógenos vía oral, o preferentemente transdérmicos, con lo que se logra el crecimiento uterino y sangrados uterinos por deprivación. Sin embargo, la administración de estrógenos no induce la ovulación10.
Niñas con un comienzo temprano del hipogonadismo, sin una exposición a gonadotrofinas, lleva a una falla en el crecimiento y desarrollo uterino, un pobre engrosamiento endometrial con una deficiente vascularización. Se describe como útero hipoplásico12.
A pesar de estas características, las mujeres que presentan hipogonadismo hipogonadotrófico aislado, luego de un tratamiento de fertilidad, tienen una alta tasa de éxito13. Concomitantemente, esta paciente presenta un déficit de GH, que fue suplementado en la infancia. Esta hormona también tiene incidencia en el desarrollo y crecimiento de los órganos reproductores. Se ha demostrado que la deficiencia y resistencia de GH modifican la foliculogénesis, maduración del ovario y ovulación14.
En esta paciente también se puede observar el compromiso de la densidad mineral ósea; se destacan dos factores que tienen una participación central: el déficit de gonadotrofinas y de GH.
Según algunos autores, el hipogonadismo congénito tiene per se aumentado el riesgo de osteoporosis, dado que el pico de masa ósea se encuentra reducido, incluso cuando se realiza el reemplazo hormonal adecuado, aunque existen controversias al respecto15,16. En esta paciente el reemplazo hormonal fue tardío, lo que podría explicar, junto con el déficit de GH, la masa ósea debajo de lo esperado para su edad.
Se conoce que las células óseas (osteoclastos, osteblastos, osteocitos) presentan receptores de estrógenos y estas hormonas tienen la capacidad de estimular la formación ósea e inhibir la resorción17,18.
Por otro lado, el déficit de GH también está asociado con baja densidad mineral ósea y recambio óseo. Por consiguiente, pacientes con déficit congénito que no reciben suplemento de GH de por vida, presentan un riesgo aumentado de fractura19. Asimismo, los pacientes que presentan déficit de GH tienen aumento de la mortalidad cardiovascular, que puede ser atribuida al incremento de lesiones ateroscleróticas. También existen cambios en el perfil lipídico, niveles elevados de triglicéridos LDL y colesterol total y bajos de HDL, sin embargo estos cambios son modestos. Estudios que comparan adultos sanos y con déficit de GH muestran que estos últimos presentan cambios en la composición corporal: aumento de la masa grasa con distribución abdominal y visceral, lo que probablemente contribuya al aumento del riesgo cardiovascular20. Por lo tanto, en pacientes con déficit congénito de GH sería beneficioso el tratamiento hormonal de por vida.
Estos pacientes con hipogonadismo tienen un riesgo elevado de presentar enfermedades cardiometabólicas, como hipertensión arterial, dislipemia, diabetes mellitus tipo 2 y obesidad, algunas de las que presenta esta paciente. Los mecanismos exactos acerca de cómo ocurre, no están completamente elucidados, pero sí hay reportes del daño endotelial, inflamación y resistencia a la insulina21,22. A su vez, estas patologías tienen incidencia negativa en los tratamientos de fertilidad23. La prevalencia de síndrome metabólico y los índices de mortalidad por causa cardiovascular están aumentados en pacientes con hipopituitarismo24.
Conclusiones
El síndrome de De Morsier es una enfermedad rara, con diversas manifestaciones y variantes. Las alteraciones faciales y oftalmológicas deben hacer sospechar el diagnóstico de esta enfermedad. El hipopituitarismo que puede acompañar este síndrome está asociado con aumento de la mortalidad, así como aumento de las enfermedades cardiovasculares y respiratorias. Es fundamental un alto grado de sospecha para así lograr un diagnóstico temprano y evaluar precozmente las alteraciones, tratarlas y realizar seguimiento en conjunto con un equipo multidisciplinario y de este modo prevenir una mayor morbilidad. La fertilidad en estas pacientes es posible mediante la inducción de la ovulación.

