











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Introducción
La displasia fibrosa (DF) es una lesión ósea benigna de baja frecuencia, congénita pero no hereditaria, debida a la mutación somática en el embrión de una célula madre y caracterizada por la proliferación medular de un tejido fibroso que encierra una osteogénesis inmadura. En ocasiones causa dolores, fracturas y deformaciones óseas1. Fue descrita por primera vez por Lichtenstein en 1938 y por Lichtenstein y Jaffe en 19421. Se puede presentar en cualquier hueso del esqueleto. Esta lesión osteofibrosa puede afectar un hueso (monostótica) o varios (poliostótica), siendo la forma monostótica de seis a diez veces más frecuente2. Se han descrito complicaciones más severas para el caso de las formas poliostóticas, obligando además a descartar las formas sindromáticas como el McCune-Albright, síndrome de Mazabraud y la displasia osteofibrosa.
El síndrome de McCune-Albright (SMA) es de muy baja frecuencia, en Uruguay no contamos con datos estadísticos. En Francia se describe una incidencia estimada de 1/100.000-1/1.000.000 habitantes3. Este síndrome se conforma por la asociación de DF poliostótica y una o más manifestaciones extraesqueléticas: lesiones hiperpigmentadas en piel (manchas color café con leche) y endocrinopatías4. De estas últimas la más frecuente es la pubertad precoz, principalmente en niñas.
El SMA resulta de mutaciones esporádicas somáticas poscigóticas en el gen que codifica la subunidad á de la proteÍna Gs (GNAS1). Esta proteÍna actúa en la transducción de senÞales mediante la unión a la adenil-ciclasa productora de adenosÍn monofosfato cÍclico (AMPc). El AMPc intracelular normalmente estimula la proliferación de las células en las glándulas (tiroides, corteza suprarrenal, ovario y pituitaria), que dan lugar a los tumores hiperfuncionantes en estos pacientes. La patogénesis de las lesiones óseas no es clara, se cree que la proteÍna Gs activada por mutaciones puede ejercer su efecto patogénico sobre los osteoclastos o fibroblastos llevando a la DF.
Es fundamental el diagnóstico oportuno y temprano de dicho síndrome para lograr un adecuado tratamiento y evitar complicaciones a corto y largo plazo. Se presenta en este estudio el caso de una niña de 8 años con DF poliostótica asociada a alteraciones endócrinas, la cual corresponde a un SMA.
Caso clínico
Escolar, sexo femenino, de 8 años, que se presenta en el servicio de traumatología con una fractura patológica de fémur izquierdo. Fractura secundaria a una caída desde su altura, mientras patinaba, quedando con dolor e impotencia funcional absoluta en miembro inferior izquierdo. Relató como único antecedente del polo óseo una cojera leve sin estudio.
Del balance lesional, presentó un síndrome fracturario de fémur izquierdo, mostró piel sana y el examen neurovascular fue normal. En la radiografía, presentó una fractura de fémur izquierdo medio diafisaria que asienta en un hueso de características patológicas (figura 1).
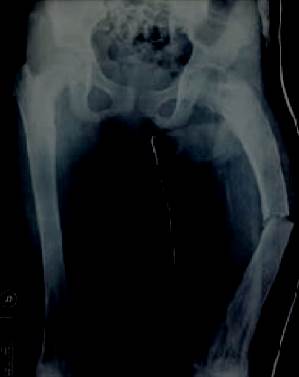
Figura 1 Displasia fibrosa en ambos fémures, con la fractura que motivó la consulta. Podemos observar la deformación de fémur proximal izquierdo. Esta deformidad en varo también es denominada en cayado de pastor (shepherd’s crook) por su similitud al mango del pastor de ovejas (izquierda).
Se evidencia un trazo de fractura transversal, medio diafisaria, desplazada, en un hueso con alteraciones en su estructura dado por imágenes radiolúcidas endomedulares que adelgazan la cortical. A su vez, se acompaña con la deformación en varo de la extremidad proximal de fémur en cayado de pastor. Estas alteraciones morfológicas nos orientan hacia a una DF. Debido a que asienta en un hueso con elementos sugestivos de un tumor benigno subyacente por presentar bordes bien delimitados, sin reacción perióstica, sin compromiso de partes blandas, que además se destaca por localizaciones múltiples, nos orientamos a DF y fibroma no osificante a confirmar por anatomía patológica. Las radiografías iniciales mostraron lesiones radiolúcidas con bordes escleróticos en diáfisis de ambos húmeros, a lo largo del fémur, la tibia y peroné izquierdos, así como el fémur y tibia derechos. Se destaca, a su vez, cartílagos de crecimiento abiertos en ambos húmeros, tibias y fémures.
La forma más rápida de determinar la distribución de la lesión en esqueleto es la gammagrafía ósea.
Este estudio evidenció lesiones poliostóticas a predominio de hemicuerpo izquierdo, como también en cráneo en región témporo-frontal, occipital izquierdo, ilíaco izquierdo, sexto y noveno arcos costales izquierdos y huesos de pie izquierdo. Las imágenes de SPECT mostraron hipercaptación en cuerpo de L3 (figura 2).
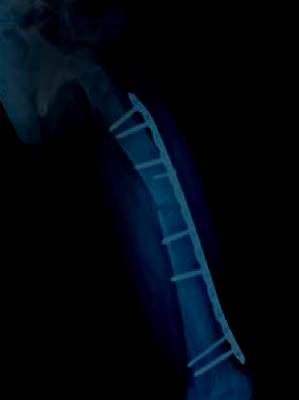
Con el planteo diagnóstico clínico-imagenológico de fractura de fémur izquierdo patológica, se realiza osteosíntesis mediante reducción abierta y fijación interna con placa bloqueada larga, al tiempo que se realiza biopsia de fragmentos óseos para el diagnóstico anatomopatológico (figura 3) y (figura 4). El resultado de anatomía patológica informa proliferación osteofibrosa constituida por células ahusadas que se disponen en un patrón fascicular, conformación de trabéculas óseas inmaduras de forma irregular sin ribete osteoblástico focal. En suma: DF.
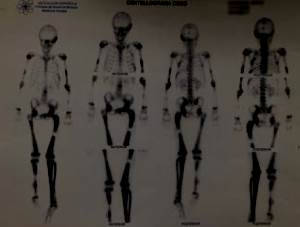
Figura 3: Centellograma óseo que evidencia lesiones poliostóticas a predominio del hemicuerpo izquierdo. Otras lesiones, además de las ya mencionadas. Las imágenes mostraron hipercaptación del radiotrazador en cráneo en región témporo-frontal occipital izquierdo e ilíaco izquierdo.
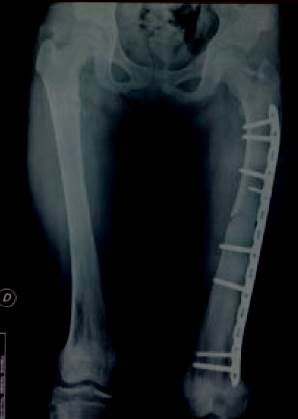
Figura 4: Radiografía control tres meses posoperatorio donde se visualiza consolidación ósea. Se presenta sin dolor y se indica apoyo total, luego de haber realizado su recuperación con muletas y apoyo parcial progresivo.
En este contexto se realizó diagnóstico clínico, imagenológico, anatomopatológico de DF poliostótica.
Del punto de vista endocrinológico la paciente presentó su menarquia a los 6 años de edad (no cíclica), luego agregó telarquia y por último la pubarquia.
Se encontró al examen físico, antropometría: talla de 135 cm (Z score: +0,75), peso de 31 kg con un índice de masa corporal (IMC) de 17 (Z score: +0,68) y una velocidad de crecimiento (Vc) calculada de 10 cm/año (>p97 para edad y sexo). A nivel de piel, no presentó lesiones hiperpigmentadas. Mamas Tanner III bilateralmente y vello púbico Tanner II, basado en la clasificación de Tanner y colaboradores en 1962 (figura 5).
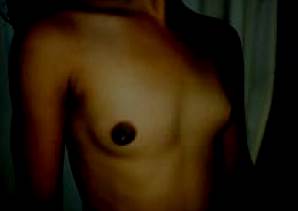
Figura 5: Mamas Tanner III: glándula mamaria que sobresale la areola, sin sobreelevación del pezón, con este último hiperpigmentado5.
La edad ósea correspondió a 12 años, para una edad cronológica de 8, lo cual se considera adelantada (> 2 desvíos estándar para edad y sexo), basado en el Radiographic Atlas of Scheletal Development of the hand and wrist de W. W. Greulich y S. I. Pyle.
De la bioquímica se destaca el perfil hormonal gonadal con valores disminuidos de la hormona folículo estimulante (FSH) y hormona luteinizante (LH) (realizado con método ICMA), con asociación de estradiol elevado (tabla 1). La dosificación de la hormona tirotropina (TSH), tetrayodotiroxina (T4), prolactina y el factor de crecimiento insulínico tipo 1 (IGF1) se encontraban dentro del rango de normalidad para la edad y el sexo.

La ecografía ginecológica informó útero AVF 53 por 28 mm, endometrio 6 mm. Ovario derecho volumen de 2 cc, ovario izquierdo volumen de 2,4 cc con dos folículos de 16 y 17 mm.
Dada la presentación clínica y paraclínica:
Pubertad precoz determinada por la aparición de caracteres sexuales secundarios a los 6 años, acompañada de:
. Edad ósea adelantada y Vc acelerada.
. Valores hormonales en sangre con estradiol alto y LH, FSH disminuidos.
. Ecografía ginecológica con características puberales y quistes ováricos mayores a 10 mm.
Corresponde a una pubertad precoz periférica, la cual asociada a la DF poliostótica, es típico del SMA.
En lo que respecta al tratamiento endocrinológico se inició letrozole 1,5 mg/m2/día.
En sexta y novena, arcos costales izquierdos y huesos de pie izquierdo. Las imágenes de SPECT mostraron hipercaptación en cuerpo de L3. (Figura 5) Mamas Tanner III: glándula mamaria que sobresale la areola, sin sobreelevación del pezón, con este último hiperpigmentado5.
Discusión
El SMA fue descrito originalmente en 1937 por la tríada clásica de alteraciones de la piel (manchas color café), pubertad precoz y DF poliostótica unilateral4,6. Luego se describieron otras patologías asociadas: hipertiroidismo, compromiso hepático, cardíaco, exceso de hormona de crecimiento y de cortisol.
Debido a la mutación del gen GNAS1, fisiopatológicamente a nivel del ectodermo afecta sobre todo a los melanocitos de la piel, en el mesodermo afecta el hueso causando alteraciones en precursores osteoblásticos y en el endodermo afecta las glándulas endócrinas, como gónadas, tiroides, hipófisis, suprarrenales, etcétera7,8. Su diagnóstico se realiza debido a la asociación de dos elementos de la tríada clásica y se confirma mediante anatomía patológica o mediante toma de ADN en sangre, piel o tejido afectado.
La DF es una lesión ósea benigna, poco frecuente. Se desconoce su verdadera incidencia, dado que existe un porcentaje considerable que son asintomáticos. A pesar de ello, se ha reportado que se encuentra entre el 5% y 7% de los tumores óseos benignos2. Dentro de estas lesiones, 50%-70% son monostóticas, 20%-30% son poliostóticas y 3%-10% presentan un SMA, revelando una muy baja frecuencia en esta patología.
Existe solo 1% de riesgo de malignidad, el cual se ha visto asociado a tratamientos previos con radioterapia, lo que obliga a un seguimiento más cercano de estos pacientes9. El pronóstico en el caso de malignización es pobre.
Las manifestaciones extraesqueléticas de DF dentro del SMA aparecen en los 3 a 10 años de edad10, que en su mayoría presentan clínica a los 5 años. En esta niña la primera manifestación extraesquelética fue su menarquia a los 6 años, por la cual no consulta médico, con progresión de su pubertad precoz.
La pubertad precoz corresponde a un pilar diagnóstico del SMA11, y se define como la presencia de caracteres sexuales secundarios en la niña antes de los 8 años, como se presentó en la paciente. La niña tenía todas las características que se describen en una pubertad de origen periférica (PPP), la cual se produce como resultado de la producción de estrógenos gonadotropina independiente, autonómica de los ovarios debido a la mutación descrita. Se diferencia de la central (PPC) por presentar primero la menarquia (no cíclica) y luego el resto de caracteres sexuales secundarios, ni ordenada, ni progresiva. Debido al aumento de estrógenos en sangre la velocidad de crecimiento se acelera (> p95 para edad y sexo) y la edad ósea suele estar adelantada. Bioquímicamente la PPP se presenta con valores de estradiol elevado en plasma y supresión de gonadotrofinas. Por último, la ecografía ginecológica puede dar un aumento del volumen de ovario unilateral y la presencia de quistes foliculares mayores a 10 mm. Todo esto se expresó en la paciente con un diagnóstico tardío.
Las manchas color café con leche se presentan en dos tercios de los SMA y hasta en un 50% de las DF poliostóticas. Corresponden al tercer pilar diagnóstico del SMA. Pueden ser una pista temprana para el diagnóstico, siendo típicamente la primera manifestación clínica. En el caso de nuestra paciente, no se encuentran dichas lesiones, pero no excluye el diagnóstico de SMA. Se caracterizan por presentar a menudo un tamaño mayor a 2 cm de bordes irregulares, recortados (tipo costa de Maine) y, por lo general, respetan la línea media.
En cuanto a otras manifestaciones extraesqueléticas de la enfermedad, se describe afectación de la glándula tiroides en dos tercios de los casos12, más frecuentemente hipertiroidismo. Se debe dosificar en sangre TSH y realizar una ecografía para descartar alteraciones estructurales. Solo la mitad de los pacientes que presentan alteraciones en la ecografía tienen un franco hipertiroidismo clínico. Es importante su diagnóstico, ya que sus alteraciones pueden avanzar la edad ósea.
Los tejidos con DF producen FGF23, lo cual lleva a la pérdida de fosfato renal, bajando el fósforo en sangre. La hipofosfaturia se relaciona con mayor índice de fractura a menor edad y además mayor dolor óseo. Su tratamiento se realiza con fosfato y vitamina D4,13, lo cual resulta en una mejora de sus síntomas. Por último, hay que descartar hiperproducción de hormona de crecimiento como, a su vez, de la prolactina, que se pueden ver en estos pacientes asociadas a tumores hipofisarios13.
En cuanto a las complicaciones óseas la fractura es frecuente y con gran índice de recurrencia, lo que repercute directamente en la calidad de vida del paciente y representa un desafío para el cirujano ortopédico en equipo multidisciplinario con endocrinología.
En cuanto a la fractura de fémur que presenta nuestra paciente, podemos decir que asienta en un hueso con alteración en su estructura y morfología. El reemplazo de hueso medular por tejido fibroso conduce a una pérdida del patrón trabecular, confiriendo en la radiografía la lesión un aspecto en vidrio esmerilado.
La deformación en cayado de pastor es característica de la DF poliostótica.
En un análisis de 46 casos, entre 1994 y 201014, se vio que existen seis tipos de deformidades femorales en pacientes con DF poliostótica, del tipo I al VI más severo. El diagnóstico suele ocurrir a los 5 años de edad. Dentro de las conclusiones, se vio que la deformación avanza a la peoría al transcurrir el tiempo. Nuestra paciente se encuentra en el tipo IV, donde se observa afectación de todo el fémur, lo cual tiene implicancias clínicas importantes, deformidad de muslo, cojera y dolor. Las fracturas de fémur suelen ocurrir complicando la situación clínica. Más allá del tratamiento quirúrgico es esperable que su deformación continúe avanzando.
En un estudio realizado por Leet A y colaboradores, publicado en 200415, con 35 pacientes, la incidencia de fractura de DF poliostótica mostró un pico entre los 6 a 10 años, que disminuye en cuanto aumenta la edad. Este estudio solo toma en cuenta las fracturas en los miembros. El sitio de mayor fractura es el fémur, seguido por húmero, tibia y antebrazo. En este estudio de 35 pacientes, 19 asociaban pubertad precoz, 9 hipertiroidismo y 7 otras endocrinopatías. Además, se encontró que 12 pacientes presentaban una fosfaturia elevada. El estudio concluye que los pacientes que presentan anormalidades metabólicas tienen fracturas precozmente. Se vio una incidencia más temprana y un aumento en la frecuencia en pacientes con fosfaturia elevada.
La consolidación ósea suele ser normal. La decisión de tratamiento dependerá del tipo de fractura, localización y deformidad ósea.
Las repercusiones endócrinas secundarias a la pubertad precoz tienen como consecuencia implicancias psicosociales, emocionales y disminución de la talla final (en algunos casos talla baja) por cierre precoz de los cartílagos de crecimiento, dado el aumento de los niveles de estrógenos en sangre. Se han planteado otros potenciales riesgos de la adultez: tendencia a la obesidad, aumentos de cifras de la presión arterial, alteración del metabolismo de hidratos de carbono y aumento del riesgo cardiovascular con incremento de la morbimortalidad, por lo que es un reto diagnóstico y es fundamental el tratamiento oportuno para el médico tratante.
Ensayos clínicos no controlados encontraron que los bifosfonatos disminuyen el dolor, mejoran la apariencia de las lesiones en las radiografías y disminuyen la incidencia de fractura, mejorando la calidad de vida16,17. Se sugiere también que la combinación de bifosfonatos e instrumentación quirúrgica puede ser beneficiosa. El uso de bifosfonatos vía intravenosa ha demostrado ser efectivo en lesiones óseas dolorosas; el uso de pamidronato parenteral ha sido reportado como que logra mejoría de la densidad ósea en forma significativa18.
Desde el punto de vista ortopédico el tratamiento de las lesiones óseas puede ser un desafío, en particular en el caso de lesiones múltiples o deformidades mayores. Sabemos que en el caso de lesiones monostóticas el tratamiento es principalmente ortopédico y solo está indicada la cirugía en caso de una gran deformidad y de un gran dolor. El tratamiento quirúrgico profiláctico con enclavijado solo es recomendado en la DF infantil. Por lo general, el tratamiento quirúrgico se indica frente a fracturas patológicas recurrentes, cuando las lesiones causan deformidad progresiva y asocian dolor persistente. Las fracturas patológicas mínimamente desplazadas consolidan en un plazo normal, por lo que el retraso de consolidación o pseudoartrosis no es un problema en estos pacientes. La principal complicación es la deformación progresiva.
Frente a una deformidad ósea el objetivo del tratamiento es realinear, particularmente en los huesos de carga. Nuestra paciente presenta una cojera debida a la desviación en varo de su cadera, por la cual está planificada su corrección quirúrgica. En niños pequeños se prefiere realizar el tratamiento ortopédico, pero en caso de fracturas repetidas en huesos largos o compromiso del fémur proximal, se prefiere el tratamiento quirúrgico.
La decisión de la técnica quirúrgica se toma con los mismos principios de osteosíntesis que en huesos no patológicos, teniendo en cuenta las alteraciones en cada caso individualizado.
El enclavijado puede ser un desafío por las deformaciones y por la calidad de hueso.
Se espera aun mayor dificultad en un esqueleto maduro. En la cirugía no se han visto beneficios al realizar curetaje, ya que éste lleva por lo general a la recurrencia local. No se recomienda utilizar injerto óseo, pues se ha observado que éste se reabsorbe y es sustituido por fibrosis nuevamente.
Leet colaboradores19. Se han visto mejores resultados con injerto óseo vascularizado20.
La decisión óptima del tratamiento quirúrgico no ha sido determinada. Hay quienes recomiendan enclavijado endomedular en lesiones en huesos de carga con DF severa porque provee estabilidad por mayor tiempo, previene nuevas fracturas y el aumento de la deformidad. El enclavijado, como contrapartida, puede lesionar la fisis y ser de difícil colocación dependiendo de la deformidad.
La decisión en nuestra paciente fue una placa bloqueada, la cual fue moldeada respetando la fisis. Se lograron los objetivos de realineación y consolidación en los tiempos apropiados (figura 3). Se va a requerir un seguimiento cercano de esta paciente, ya que sabemos que la osteosíntesis soluciona el problema agudo de la fractura, pero no la enfermedad de base.
En la evolución, osteotomías pueden ser necesarias en caso de consolidación viciosa o si está presente la deformación en fémur proximal en cayado de pastor, como en el caso de nuestra paciente, que presenta una leve cojera que se espera evolucione a la peoría en el caso de no tratarla.
Por último, en lo que respecta al tratamiento de la pubertad precoz periférica, es inicialmente el seguimiento clínico. De ser progresivo, como es el caso de la niña analizada, se usan fármacos inhibidores de la aromatasa con acción antiestrogénica (letrozol)21. Estas niñas en la adultez pueden tener comprometida la fertilidad. La descendencia no tiene por qué verse afectada.
Conclusión
Estamos frente a una niña de 8 años que presenta SMA. Es fundamental el diagnóstico temprano y el adecuado abordaje multidisciplinario. El diagnóstico de su enfermedad fue a partir de una fractura patológica de fémur izquierdo. Desde el punto de vista traumatológico, la niña es tratada mediante osteosíntesis de fémur con placa bloqueada lateral. Presenta una buena evolución clínica imagenológica con un buen proceso de consolidación ósea en los tiempos esperados, sin dolor, y vuelta a la actividad diaria. Su pronóstico funcional es bueno, siendo probablemente necesaria una nueva intervención a futuro debido a la deformación en varo de su cadera. Se restringe la actividad deportiva y se actúa en la prevención de nuevas fracturas. La paciente se encuentra en control y tratamiento con endocrinólogo por su pubertad precoz, seguimiento de repercusiones en el metabolismo fosfo-cálcico y posible aparición de otras patologías endócrinas.

