











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Rol de la dieta en el síndrome metabólico
Los cambios en la composición de la dieta asociados con la dieta occidental contribuyen en gran medida a las alteraciones bioquímicas que promueven el síndrome metabólico, entidad que ha adquirido proporciones epidémicas1-3. Descrito por Gerald Reaven hace casi cinco décadas, tiene clásicas definiciones diagnósticas muy conocidas por el cuerpo médico y remitimos al lector a excelentes revisiones previas4,5. Con un encare funcional y mecanístico nos permitimos definir el síndrome metabólico como una constelación de hallazgos fisiopatológicos enmarcados en un aumento de grasa ectópica (o sea en el hígado, epiplón, mesenterio y en el músculo), inflamación, insulino-resistencia (IR), disfunción hepática y defectos en el tráfico de glucosa y lípidos(4, 5). La esteatosis hepática no alcohólica, la diabetes tipo 2 y la aterosclerosis son algunas de las morbilidades asociadas con el síndrome (figura 1). Los componentes genéticos del síndrome metabólico son bien conocidos, sin embargo, la dieta, un componente del estilo de vida, juega un papel importante en esta epidemia4,6-8. Clásicamente los lípidos saturados y los trans, así como los aminoácidos de cadena ramificada, han sido vinculados a la esteatosis hepática, a la cual muchos autores consideran causal en la IR hepática, que consecuentemente produce la IR sistémica como corolario9,10. Sin embargo, la fructosa es un nutriente causal aún más importante ya que: (a) su consumo en todas las formas, especialmente en forma líquida, ha aumentado significativamente junto con la incidencia de condiciones de síndrome metabólico, (b) más del 80% de la fructosa ingerida se metaboliza en el hígado en el primer paso, donde estimula la lipogénesis de novo (DNL, por su sigla en inglés), o sea la producción de triglicéridos hepáticos (TG) que (c) contribuye a la esteatosis hepática no alcohólica, la resistencia a la insulina hepática y la dislipidemia11,12. En última instancia, el síndrome metabólico es la expresión fenotípica de la IR, se asocia con la obesidad y ambos están adquiriendo prevalencias elevadísimas en las últimas décadas.
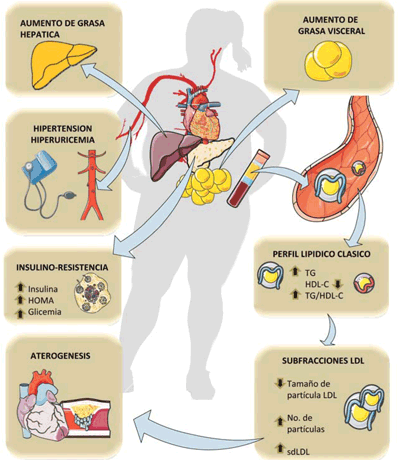
Figura 1 Figura 1: Principales componentes del síndrome metabólico y su relación con el consumo de fructosa que se exploran en esta revisión. HOMA: Homeostatic Model Assessment (valores altos indican resistencia a la insulina); sdLDL: LDL pequeña y densa. Diagrama realizado en parte utilizando Servier Medical Art https://smart.servier.com/
Mala ciencia e intereses económicos nos atrasaron varias décadas
Hace un siglo, en Europa ya se había mostrado la correlación del consumo de azúcar con diversas enfermedades metabólicas. En la década de 1960, Yudkin, en el Reino Unido, había logrado pruebas contundentes sobre su rol en la aterogénesis y obesidad13,14. Sin embargo, Allen Keys15-17, en Estados Unidos, un epidemiólogo estrella, basado en a) estudios epidemiológicos con un sesgo importante; b) el razonamiento pueril de que los lípidos y el colesterol de la dieta son los mismos que se acumulan en las arterias; c) presiones del lobby del maíz; d) la necesidad de Richard Nixon de abaratar los alimentos para favorecer su reelección (subvencionando el maíz) logra que, en la década del 1980, las recomendaciones del Instituto Nacional de la Salud (NIH) en Estados Unidos culparan a los lípidos en el desarrollo de la aterosclerosis y de la obesidad, lo que nos enseñaron y enseñamos luego en el área de la salud y se transformó en dogma. Se cambió la dieta estadounidense: se bajó el consumo de grasa aumentándose el de almidón y azúcar con el resultado flagrante (y opuesto al postulado) de un incremento en proporciones inéditas del riesgo cardiovascular, obesidad, síndrome metabólico y diabetes mellitus de tipo 2 en cuatro décadas1,18,19. El poder del “establishment” es tal que se necesitó llegar al 2015 para que la American Heart Association se decidiera a eliminar el colesterol de la dieta como factor de riesgo cardiovascular. Y la evidencia sobre el rol de las grasas saturadas aisladamente tampoco es convincente12. En la actualidad el péndulo oscila nuevamente hacia la fructosa, que retorna al eje de discusión, ya que una gran cantidad de evidencia muestra que la fructosa del azúcar, si es consumida en exceso y sin fibra, contribuye en gran medida a estas enfermedades metabólicas. Sin embargo, existe el falso concepto tanto en el ambiente médico, y por supuesto más aún en la población general, de que los efectos del azúcar se deben simplemente al exceso calórico, es decir, es lo mismo almidón, azúcar o grasa, solo importan las calorías totales de cada ingesta. Sin embargo, la evidencia es cada vez más clara en adjudicar un papel clave del metabolismo hepático de la fructosa -que conduce a la acumulación de grasa hepática y visceral- como un factor clave que produce IR hepática. Esta última lleva al síndrome metabólico y termina en obesidad o diabetes tipo 2. Por lo tanto, el propósito de esta revisión narrativa es destacar en forma sucinta los efectos metabólicos específicos del consumo de fructosa (más allá del contenido calórico) en el desarrollo del síndrome metabólico, la esteatosis hepática no alcohólica y su asociación con la obesidad.
Fuentes de fructosa
La fructosa es un monosacárido que se encuentra principalmente en el azúcar (sacarosa: 50% de glucosa y 50% de fructosa), frutas, miel, así como en formas procesadas como jarabe de maíz con alto contenido de fructosa y bebidas azucaradas18,20. Los estudios epidemiológicos han relacionado el consumo de fructosa (en forma de azúcar o jarabe de maíz) con la obesidad, diabetes, síndrome metabólico y esteatosis hepática21,22. La correlación con las bebidas azucaradas es particularmente fuerte18,23. El mecanismo de cómo la fructosa participa en esas patologías aún no está completamente claro, sin embargo, diferentes estudios en animales y humanos han permitido el desglose de sus efectos metabólicos más significativos, de los cuales resumimos los más importantes a continuación.
Digesto-absorción de la fructosa, rol del intestino
La fructosa libre se absorbe directamente en la luz intestinal, mientras que de la sacarosa se obtienen glucosa y fructosa por digestión. La fructosa es absorbida principalmente a través del transportador de glucosa 5 (GLUT5) por difusión en el lado luminal y a través del transportador de glucosa 2 (GLUT2) en el lado basolateral del enterocito. La fructosa ingresa al hígado desde la circulación portal, el hígado contiene dos transportadores de glucosa (a saber, GLUT 1 y 2) y dos de fructosa (GLUT 2 y GLUT 8, respectivamente). Es en el hígado donde la fructosa se metaboliza principalmente (más del 80% es extraída en el primer paso). Si se consume en forma aislada (muy raramente, como veremos) aproximadamente el 50% se convierte en glucosa, el 15%- 20% en glucógeno hepático y el 15%-25% en lactato o ácidos grasos (AG) que se secretan como triglicéridos de VLDL (lipoproteína de muy baja densidad, precursora de la LDL) o se almacenan como grasa intrahepática24,25.
El metabolismo hepático de fructosa promueve la lipogénesis
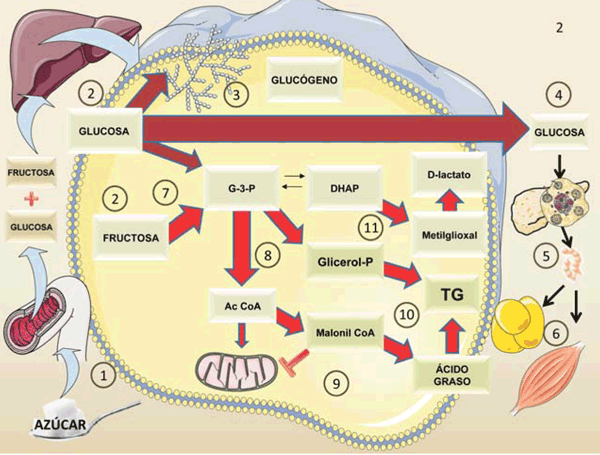
figura 2 Figura 2: Diferencias metabólicas entre la glucosa y la fructosa. No se trata solo de calorías ni de todos los carbohidratos, sino de uno específico. La figura resume en forma esquemática el metabolismo de 100 g de azúcar (50% de glucosa y 50% de fructosa). Después de la digestión del azúcar:
1) la glucosa y la fructosa ingresan a la vena porta en cantidades iguales 2) En el hígado, la glucosa se convertirá en glucógeno 3), la mayor parte pasa al torrente sanguíneo 4) para alimentar los tejidos a medida que aumenta la secreción de insulina 5) y la glucosa ingresa a los músculos y tejido adiposo 6) La fructosa no abandona el hígado en su mayor parte. En cambio, es rápidamente fosforilada por la fructoquinasa C, evitando los pasos reguladores en la glucólisis e inundando el sistema 7) y 8). Los metabolitos atrapados tienen un solo destino posible, convertirse (directa o indirectamente) en grasa por lipogénesis de novo 9) y 10). El proceso disminuye la oxidación de AG por la mitocondria, ya que el malonil CoA inhibe el transporte de AG a la mitocondria 9) Las triosas también se transforman en el metabolito tóxico metilglioxal, que puede detoxificarse a D-lactato 11) Estos procesos tienen consecuencias deletéreas, como se explica en las siguientes figuras. Compárese con la figura 3. Diagrama realizado en parte utilizando Servier Medical Art https://smart.servier.com/
En el hígado, tres enzimas clave metabolizan la fructosa. Primero, la fructosa se fosforila a fructosa 1 fosfato (fructosa-1-P) por la enzima fructoquinasa C (FFK C), también llamada cetohexoquinasa (KHK). La fructosa-1-P luego se convierte en dihidroxiacetona-fosfato (DHAP) por la enzima aldolasa B y en gliceraldehído-3-fosfato (G-3-P) a través de la trioquinasa. Estas triosas participan en otras vías metabólicas: glucólisis, síntesis de lípidos, gluconeogénesis y glucogénesis (figura 2) y (figura 3). Es muy importante tener en cuenta que la fructosa saltea las dos etapas claves reguladas de la glucolisis (hexo o glucoquinasa y la fosfofructoquinasa) y, en consecuencia, usa pasos no regulados y esto permite flujos metabólicos deletéreos cuando hay exceso de sustrato 20,24,25.
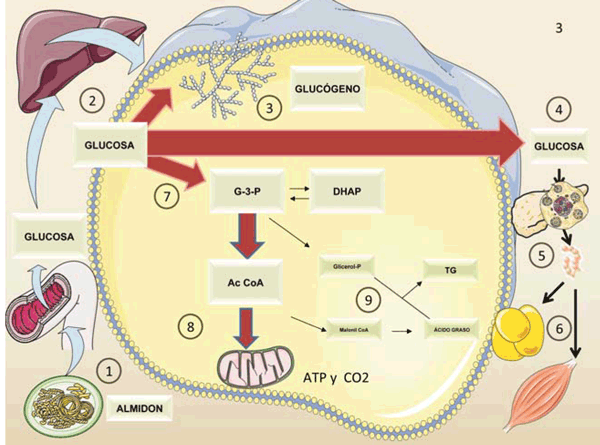
Figura 3 Figura 3: Diferencias metabólicas entre la glucosa y la fructosa. La figura resume en forma esquemática el metabolismo de 100 g de glucosa (del almidón). Después de la digestión del almidón:
1) la glucosa entra en la vena porta 2) En el hígado se convertirá, en parte, en glucógeno 3), la mayor parte irá al torrente sanguíneo 4) para alimentar los tejidos a medida que aumenta la secreción de insulina 5) y la glucosa ingresa a los músculos y tejido adiposo 6). La glucólisis 7) produce piruvato y luego AcCoA mitocondrial 8) Muy poca AcCoA sale de la mitocondria (a través del citrato) para convertirse en grasa 9), mediante el proceso llamado lipogénesis de novo (DNL). Compárese con la figura 2. Diagrama realizado en parte utilizando Servier Medical Art https://smart.servier.com/
Fructosa y glucosa: la gran diferencia
Como adelantamos, la mayor parte de la fructosa ingerida se extrae de la sangre portal a través del metabolismo hepático de primer paso, mientras que solo una pequeña fracción de la fructosa ingerida eventualmente ingresará a la circulación sistémica. Pero es necesario decir que raramente consumimos fructosa aislada, sino que se ingiere con glucosa (en azúcar o jarabe de maíz). La mezcla explosiva resultante de un flujo portal de glucosa -potente secretagogo de insulina- y de fructosa que no lo es, hace toda la diferencia. Para resaltar mejor dichas diferencias, comparemos, como se ilustra en las figuras 2 y 3, lo que sucede con una carga de glucosa (100 g de pasta, por ejemplo, figura 3) y la misma carga de sacarosa (fructosa y glucosa en 120 g de dulce o 1 litro de bebida azucarada, figura 2). La glucosa es usada para formar glucógeno y proveer glucosa a la circulación en su mayoría. La fructosa no tiene otro destino en esas circunstancias (porque la glucosa es abundante y ha estimulado un pico de insulina) que promover la lipogénesis no solamente suministrando sustrato, sino además activando la proteína de unión al elemento de respuesta a carbohidratos (ChREBP, por su sigla en inglés)26,27.
Estas afirmaciones están corroboradas por muchos estudios en animales y humanos que han demostrado que la fructosa, en comparación con la glucosa o el almidón en las dietas con la misma cantidad de calorías, aumenta la grasa visceral, los TG circulantes y la presión arterial21,22. Ergo, no todas las calorías son iguales fisiológicamente. Todas estas son las características fisiopatológicas del síndrome metabólico.
Si se nos permite un símil para ilustrar el concepto, piénsese en el “abdomen de cerveza”. En ese caso hay aumento de grasa visceral y esteatosis hepática porque la glucosa de la cerveza (la maltosa es un disacárido de glucosa) es secretagoga de insulina y porque el alcohol se transforma en acetato (acetil-CoA) que suministra los carbonos y NADPH que proporciona el poder reductor para la síntesis de ácidos grasos. Si cambiamos el vocablo alcohol por fructosa, el resultado final es el mismo. La acumulación de grasa en el hígado es el enlace clave con la IR. En efecto, existe una estrecha relación entre el consumo de fructosa, lipogénesis y esteatosis hepática. Como se mencionó previamente, la fructosa aumenta la FFK C hepática e induce la lipogénesis que predispone a esteatosis hepática, un proceso caracterizado por un desequilibrio entre los TG sintetizados a través de la lipogénesis o de los ácidos grasos reciclados de la lipólisis del adipocito por un lado, y su oxidación o exportación como VLDL desde el hígado, por otro. El exceso de grasa en el hígado puede conducir al desarrollo de IR hepática, que es clave en el síndrome metabólico, así como a la esteatohepatitis no alcohólica, una etapa que predispone a la cirrosis y sus complicaciones7,9. El mecanismo preciso está aún en discusión, pero es claro que el diacilglicerol (etapa anterior a formar un TG) y las ceramidas (lípidos complejos) inhiben la actividad del receptor de la insulina al promover la fosforilación del sustrato del receptor de la insulina (IRS 1) en una serina en lugar de una tirosina10. De otra manera, cuando se aumenta el flujo de sustrato para producir grasa, estos metabolitos intermedios se acumulan inevitablemente y disminuyen la señalización de la insulina.
Además, se ha propuesto que ese mismo aumento en el flujo de triosas también aumenta la generación de metilglioxal (MG), un dicarbonilo muy tóxico y su producto de desintoxicación, D-lactato28,29. El estrés de dicarbonilo promovido por el MG (que daña muchas proteínas) puede desempeñar un papel importante en el desarrollo del síndrome metabólico30. Hemos mostrado que en adolescentes obesos, el D-lactato es 40% más alto que en los de peso normal, se asocia con dislipidemia y que la restricción de fructosa lo reduce en paralelo con la reducción de la lipogénesis y la grasa hepática29,31. Por lo tanto, propusimos que la fructosa puede jugar un papel clave en la obesidad y el síndrome metabólico por este nuevo mecanismo que se agrega a los descritos previamente.
La importancia del metabolismo de la fructosa en la NAFLD no puede quedar más clara si observamos que Pfizer está desarrollando (fase 1) PF-06835918, un inhibidor de la FFK C32.
La fructosa aumenta la formación de ácido úrico y tiene efectos hipertensivos
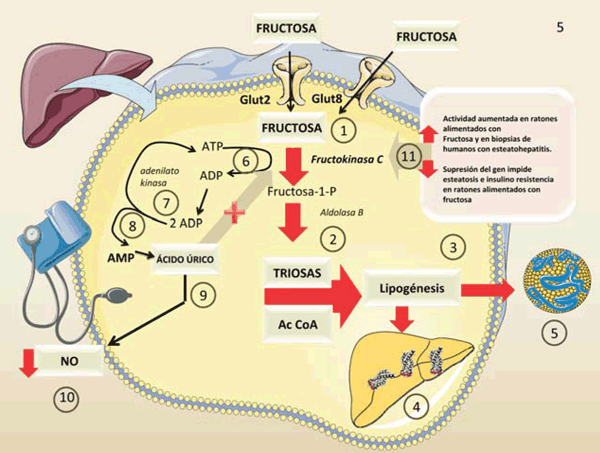
figura 4 Figura 4: Fructosa y ácido úrico. Rol hipertensivo. Fructosa y ácido úrico. Rol hipertensivo. En el hígado la FFK C activa: 1
) inunda el citosol con triosas y AcCoA 2), lo que conduce a lipogénesis 3), hígado graso 4) e hiperlipidemia 5), especialmente porque la fructosa se ingiere conjuntamente con la glucosa que se usa para la producción de glucógeno. Esto impulsa la secreción de insulina que aumenta la lipogénesis 3). La fosforilación rápida y no regulada de la fructosa conduce a un rápido consumo del ATP citosólico 6). Para reponer el ATP citosólico, las células usan adenilato quinasa 7) para generar 1 ATP y 1 AMP a partir de 2 ADP 8)ElAMP es un producto final que se degrada en ácido úrico. El ácido úrico disminuye el NO, lo que conduce a un deterioro del tono vascular e hipertensión
El ácido úrico, metabolito final del AMP generado por el metabolismo de la fructosa, activa el sistema renina-angiotensina, inactiva (químicamente formando 6-aminouracilo) el vasodilatador óxido nítrico endotelial (NO) y disminuye su síntesis, lo que provoca un aumento de la presión arterial que es la otra carta de presentación del síndrome metabólico33-35. Los flujos de ácido úrico inducidos por la fructosa son el resultado de deficiencias de energía transitorias generadas por la fosforilación rápida no regulada de la fructosa, como se muestra en la figura 2. En la figura 4 ampliamos los detalles de este proceso.
Una característica importante del ácido úrico, que potencia el problema, es que se ha demostrado que es un activador de FFK C y, por lo tanto, un perpetuador de este ciclo. En realidad, la mutación por la que hemos perdido la capacidad de metabolizar el ácido úrico durante la evolución se ha propuesto como una característica evolutiva ventajosa, que facilita la asimilación de frutas en tiempos de abundancia. La expresión de la uricasa en animales experimentales reduce los efectos nocivos de la fructosa. La importancia de la FFK C se evidencia aun más en estudios recientes que demuestran que su actividad aumenta tanto en humanos obesos con NASH como en ratones alimentados con fructosa32,36. Su inactivación previene la esteatosis inducida por fructosa y la IR. Diversos estudios han informado que el ácido úrico induce estrés oxidativo e inflamación, aumentando la lipogénesis32. Se ha demostrado con estudios del inhibidor de la síntesis de ácido úrico, como el alopurinol, que el ácido úrico favorece la esteatosis por efecto del aumento de la lipogénesis37. La crítica interacción de los metabolismos de la fructosa, el ácido úrico y el síndrome metabólico es tan clara que se están desarrollando nuevos inhibidores para atacar ambos problemas32,37.
Visión panorámica del metabolismo de la fructosa
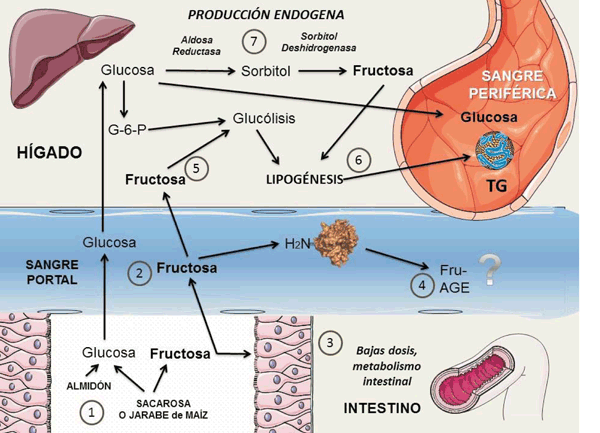
figura 5 Figura 5: Visión panorámica del metabolismo de la fructosa:
1) Lafructosa, en su mayoría ingerida en forma de sacarosa o jarabe de maíz, se absorbe en su casi totalidad y termina en el hígado por vía porta 2). A bajas dosis es posible que el intestino actúe como buffer 3). La fructosa es muy activa en la reacción de Maillard y puede formar productos de glicación avanzada (AGE) 4). El metabolismo hepático no regulado 5) favorece la lipogénesis 6). Puede haber una fuente extra de fructosa, la fructosa endógena 7), por la vía de los polioles, y a partir de glucosa. Diagrama realizado en parte utilizando Servier Medical Art https://smart.servier.com/
Como vimos en las (figuras 2y3), la fructosa, en su mayoría ingerida en forma de sacarosa o jarabe de maíz, se absorbe y termina en el hígado por vía porta. A bajas dosis (e.g: ingesta de una o dos porciones de fruta), es posible que el intestino actúe como buffer38. En efecto, recientemente estudios en ratones muestran que las dosis muy bajas de fructosa son captadas en un 90% por el intestino. Las altas dosis de fructosa (e.g: medio litro de gaseosa o jugo de naranja) abruman la absorción y el metabolismo de fructosa intestinal, lo que da como resultado que la fructosa llegue a la microbiota del colon por un lado y al hígado por otro. De esta manera el intestino delgado protegería al hígado de la exposición a la fructosa que de otro modo es nociva, como destacamos38. Otra fuente de fructosa que parece cada vez más importante es la producción endógena a partir de exceso de glucosa, por la vía de los polioles, muy conocida por los diabetólogos. Como se muestra en la figura 5, la aldosa reductasa (AR) no es muy activa a niveles normales de glucosa. Pero en condiciones de hiperglicemia, los niveles de glucosa en los tejidos independientes de la insulina (hígado, endotelios, epitelio renal, etc.) aumentan y, en consecuencia, se activa la vía del poliol. La glucosa se transforma en sorbitol. La sorbitol deshidrogenasa (SDH) actúa sobre el sorbitol, lo que conduce a la formación de fructosa, utilizando reductores como el glutatión. Un flujo excesivo por esta vía puede así afectar potencialmente el equilibrio redox en la célula39. Cada vez hay más pruebas de que la aceleración de la vía del poliol está implicada en la patogénesis de las complicaciones vasculares diabéticas (a causa de la hiperglicemia) y más recientemente que la producción endógena de fructosa a nivel del hígado no es despreciable39.
Fructosa y diálogo cruzado entre el tejido adiposo visceral y los hepatocitos: el papel del cortisol
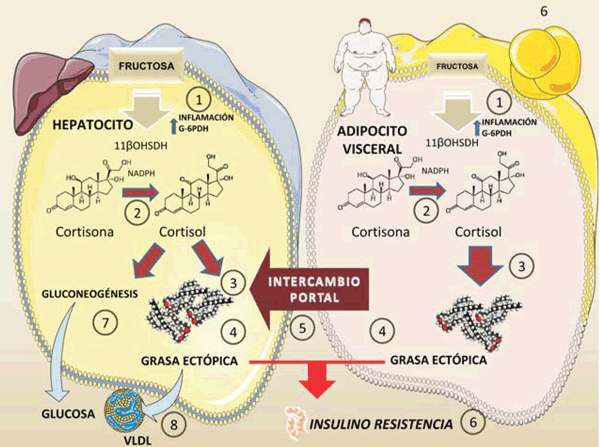
figura 6 Figura 6: Fructosa y diálogo cruzado entre el tejido adiposo visceral y los hepatocitos. El papel del cortisol. La fructosa en los hepatocitos (más del 80% de la ingesta) o el tejido adiposo visceral (concentraciones mucho más bajas pero no insignificantes) induce inflamación local y activación de G6PDH,:
1). Esto conduce a la activación de 11-beta-OHSDH que convierte en cortisol a la cortisona inactiva 2). El cortisol (efecto crónico) estimula la síntesis de grasa, 3) en ambos tejidos, así como el depósito de grasa ectópica 4). El tejido adiposo visceral envía a través de la circulación portal moléculas inflamatorias (Il-6, resistina etc.) y ácidos grasos, lo que aumenta la IR del hígado. El cortisol estimula la gluconeogénesis y la producción de glucosa hepática, 7) así como la hiperlipidemia 8). Como se ve, todos estos efectos se superponen y son sinérgicos con los anteriormente resumidos. Diagrama realizado en parte utilizando Servier Medical Art https://smart.servier.com/
La activación de glucocorticoides inactivos, como la cortisona en humanos y la 11-deshidrocorticosterona en roedores, a sus formas activas, cortisol y corticosterona, respectivamente, produce un aumento en la biodisponibilidad de estas formas activas dentro de las células. Esta transformación es catalizada por la enzima 11 beta-hidroxiesteroide deshidrogenasa (11-beta-OHSDH), que se expresa tanto en el hígado como en el tejido adiposo40,41.
Esta enzima es crucial para la activación de glucocorticoides y depende del nicotinamida adenina dinucleótido fosfato (NADPH). La fructosa, justamente, activa los glucocorticoides porque estimula a la vez un estado inflamatorio y aumenta el NADPH, que a su vez induce la 11-beta-OHSDH. Además, se ha demostrado que la fructosa es capaz de afectar la expresión génica de 11-beta-OHSDH isoforma 140,41.
Los pacientes con síndrome metabólico muestran una mayor expresión de 11-beta-OHSDH y cortisol intracelular, en forma similar al síndrome de Cushing, que se caracteriza por un exceso de glucocorticoides y es capaz de producir por sí mismo síndrome metabólico y diabetes tipo 2.
En conclusión, la 11-beta-OHSDH aumenta el cortisol intracelular (que puede ser incrementado por la fructosa), lo cual aumenta la grasa visceral, inflamación, IR, hiperlipidemia e hipertensión, características del síndrome metabólico.
Fructosa, sobrepeso y obesidad
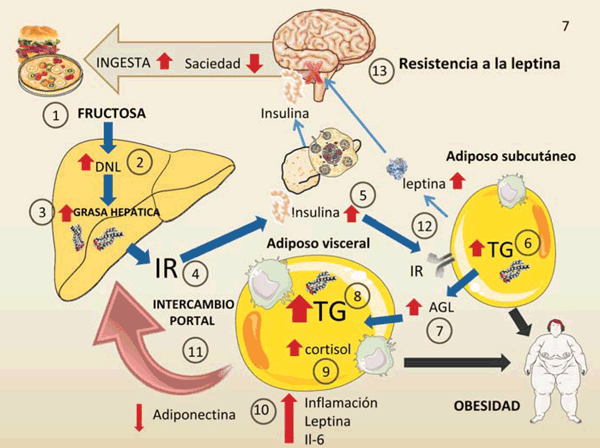
figura 7 Figura 7: Principales vías del metabolismo de la fructosa que conducen a resistencia a la insulina, síndrome metabólico y obesidad. Este diagrama resume los mecanismos claves a nivel sistémico. Las oleadas de fructosa (especialmente en forma líquida):
1) (junto con glucosa que aumenta la secreción de insulina) aumentan la lipogénesis 2) y la grasa hepática 3). Estos a su vez generan IR hepática 4). La hiperinsulinemia se produce como un mecanismo de compensación 5). La grasa subcutánea, menos resistente a la insulina, acumula TG 6) pero también aumenta la producción de ácidos grasos 7). La grasa visceral capta los ácidos grasos y acumula TG 8), aumenta de tamaño y se inflama, hace hipoxia, muere, atrae macrófagos y genera inflamación. La producción in situ de cortisol potenciada por la fructosa incrementa este efecto 9). La grasa visceral vuelca sus moléculas inflamatorias y ácidos grasos libres en la circulación portal 10), lo que aumenta la IR hepática 11). La grasa subcutánea y visceral aumenta la secreción de leptina, lo que llevaría a una disminución del apetito y a un mayor gasto de energía 12) Sin embargo, la hiperinsulinemia conduce a una resistencia a la leptina del sistema nervioso central. Esto conduce a menos saciedad, aumenta ingesta de alimentos y el ciclo continúa. DNL: lipogénesis de novo; AGL: ácidos grasos libres. Diagrama realizado en parte utilizando Servier Medical Art https://smart.servier.com/
Más del 80% de los estudios de autores que declaran no tener conflicto de intereses con la industria del azúcar o los lobbies políticos, encuentran una correlación positiva entre las bebidas azucaradas (gaseosas, jugos de fruta y bebidas deportivas) y la obesidad22. La forma en que la fructosa aumenta el apetito o disminuye la saciedad es induciendo una doble resistencia a la insulina y leptina, como se muestra en la figura 7. La leptina es una hormona sintetizada principalmente en el tejido adiposo, que circula en proporción a la grasa corporal. Es un regulador clave de la ingesta de energía a través de su interacción con los centros hipotalámicos, lo que aumenta a la vez la saciedad y el gasto energético. Sin embargo, tanto la obesidad como el consumo de azúcar (por la hiperinsulinemia) inducen una resistencia a la leptina en los centros hipotalámicos, por lo tanto, la respuesta de saciedad que se debe producir es inhibida, lo que resulta en un mayor consumo de alimentos42. El consumo crónico de fructosa se asocia con un aumento de leptina en plasma42-44. La fructosa también tiene un efecto sobre las hormonas intestinales relacionadas con la saciedad, por falta de inhibición de la liberación de grelina desde el estómago, lo que produce un efecto orexigénico. Además, reduce las hormonas de saciedad como el péptido similar al glucagón 1 (GLP-1) y el péptido YY (PPY). Otro mecanismo involucrado en el aumento de peso asociado con el consumo de fructosa podría ser el efecto que tiene el azúcar en inducir respuestas hedónicas al estimular la dopamina en el núcleo accumbens y en el mesencéfalo. Una estimulación repetida de la dopamina por el azúcar podría alterar la función de sus receptores; esto se ha demostrado en sujetos obesos a través de estudios de imagenología, mientras que los estudios en animales muestran signos de abstinencia al eliminar el azúcar26,27.
Por lo tanto, el aumento de peso y la obesidad inducida por la fructosa podrían estar relacionados con la respuesta adictiva al consumo de azúcar, la reducción de ATP en el hígado y la promoción de la resistencia a la leptina.
Como corolario de los diferentes procesos descritos en esta revisión nos queda claro que el metabolismo de grandes dosis de fructosa (recordemos que 1 litro de jugo de frutas o de bebidas azucaradas, por ejemplo, contiene 60 g o más, acompañada de otros 60 g de glucosa que es el secretagogo de insulina más importante), favorece la lipogénesis, la esteatosis hepática y la IR. El resultado crónico es el síndrome metabólico y la diabetes tipo 2.
Nuestros aportes para atacar el problema
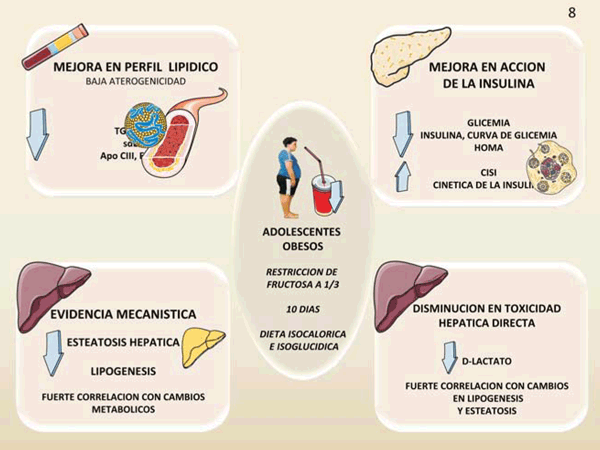
figura 8 Figura 8: Nuestro aporte al tema: profundos cambios metabólicos luego de restricción de fructosa. La figura resume en forma esquemática nuestros estudios. La restricción de fructosa con sustitución isocalórica de carbohidratos complejos en niños obesos cuyas dietas habituales eran altas en azúcar dio como resultado: descenso de TG, LDL-C, sdLDL (LDL pequeña y densa); mejora de la glucosa en ayunas, insulina y su cinética, mejora de la curva de glicemia a; disminución de la lipogénesis hepática; disminución de la grasa hepática y disminución del D-lactato. CISI: Composite Insulin Sensitivity Index (valores altos indican mayor sensibilidad a la insulina): HOMA: Homeostatic Model Assessment (valores altos indican resistencia a la insulina). Diagrama realizado en parte utilizando Servier Medical Art https://smart.servier.com/
Todas estas observaciones, que objetivan la relación entre el consumo de fructosa y el síndrome metabólico, obesidad, esteatosis hepática, activación de corticosteroides y producción de MG, son en la actualidad objeto de intenso escrutinio científico. La Organización Mundial de la Salud y la American Heart Association (AHA) sugieren limitar el consumo de azúcar a niveles que son la tercera parte del consumo habitual en Estados Unidos, por ejemplo, a los que se agregan las recomendaciones en Uruguay1,22. Sin embargo, existe una corriente que niega los efectos específicos de la fructosa y adjudica todo el problema a un aumento en la ingesta calórica. Como ocurrió hace cinco décadas con el tabaco (45, la industria financia investigación que crea dudas46,47. Lo que hoy resulta obvio con el cigarrillo será moneda corriente en el futuro cuando el peso de la evidencia de la falacia de las calorías sea abrumador. Pero antes, habrán muerto millones de personas por causas que se pueden prevenir.
Precisamente, para probar esta especificidad de la fructosa se necesitan estudios de intervención en humanos que ayuden a establecer la veracidad de los mecanismos propuestos. Dado que la sobrealimentación de los humanos con fructosa (azúcar) llevaría a un aumento de peso, hemos realizado estudios de reducción de la ingesta de fructosa (en aproximadamente 2/3) en adolescentes obesos durante solo diez días, como resumimos en la figura 8. La restricción de fructosa con sustitución isocalórica de carbohidratos complejos en adolescentes obesos cuyas dietas habituales eran altas en azúcar dio como resultado: descenso de triglicéridos, LDL colesterol, LDL pequeña y densa (sdLDL, la partícula realmente aterogénica), mejora de la glucosa en ayunas, de la insulina y su cinética, mejora de la curva de glicemia; disminución de la lipogénesis hepática; disminución de la grasa hepática y disminución de D-lactato31,48-50.
Estos resultados sugieren que la lipogénesis hepática es un mecanismo importante que contribuye a la acumulación de grasa hepática ya en los adolescentes, lo que puede revertirse por la restricción de fructosa a corto plazo. Estos datos fortalecen el papel protagónico de la fructosa en el síndrome metabólico: la fructosa incrementa la grasa hepática, que a su vez produce la IR que inicia el ciclo patogénico.
Conclusión
En esta revisión hemos mostrado el rol específico importante que el exceso de consumo de fructosa puede tener en el desarrollo del síndrome metabólico, la esteatosis hepática no alcohólica y su asociación con la obesidad más allá de sus calorías. Se resumen los efectos específicos de la fructosa (en comparación con la glucosa) en la esteatosis hepática. La evidencia actual subraya la necesidad de intervenciones clínicas y educativas hacia la población para regular o reducir el consumo de fructosa, especialmente en niños y adolescentes, los principales consumidores de fructosa, que han demostrado, en edades tan tempranas, alteraciones metabólicas significativas relacionadas con este consumo.

