











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Introducción
El síndrome de Budd-Chiari (SBC) se define como la obstrucción al flujo de salida venoso hepático, que puede localizarse a cualquier nivel desde las pequeñas vénulas hepáticas hasta la entrada de la vena cava inferior (VCI) en la aurícula derecha1. El SBC es una patología rara que se asocia frecuentemente a un estado protrombótico. El cuadro clínico del SBC puede ser variable. Existen casos fatales por fallo hepático grave o complicaciones relacionadas a hipertensión portal. El diagnóstico y tratamiento oportuno del SBC es crucial para mejorar su pronóstico, lo que representa un verdadero reto para el médico clínico.
A continuación se reporta un caso clínico de SBC secundario a síndrome antifosfolípido (SAF).
Caso clínico
Se presenta el caso de una mujer de 31 años derivada a la Clínica Médica “1” del Hospital Maciel, con historia de ascitis de seis años de evolución, refractaria al momento del ingreso. Repercusión general en el último año. Antecedentes personales: anafilaxia a los medios de contraste, hepatitis aguda por virus A en la infancia. Sin exposición a hepatotóxicos. Sin antecedentes familiares relevantes.
Examen físico: lúcida, con desnutrición proteico calórica severa, ictericia universal, hepatomegalia dolorosa, esplenomegalia y ascitis a tensión. Resto del examen físico sin alteraciones.
Analítica sanguínea: los resultados se exponen en la (tabla 1). En el hepatograma destacaba un patrón colestásico dado por el aumento de los valores de gamma -glutamil transpeptidasa, fosfatasa alcalina y bilirrubina total (a expensas de la bilirrubina directa). Transaminasas y albuminemia normales. Colinesterasa y tasa de protrombina descendidas, con un INR discretamente aumentado.
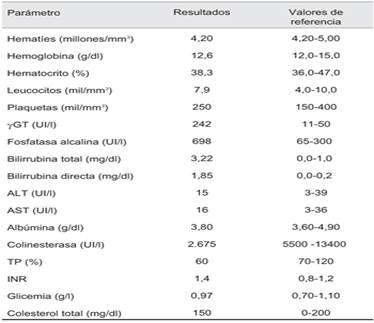
Estudios dirigidos a la búsqueda etiológica: marcadores virales de hepatitis B y C negativos; autoanticuerpos para hepatopatía autoinmune (anticuerpos antinucleares (ANA), anticuerpos de tipo 1 microsomales de hígado y riñón (anti-LKM-1), anticuerpos antimitocondriales (AMA), anticuerpos antimúsculo liso (ASMA), anticuerpos antiantígeno soluble hepático (anti-SLA)) negativos; factor reumatoideo negativo, complementemia y ferritina normales; ceruloplasmina, cupremia y excreción urinaria de cobre en 24 horas normales.
Análisis citoquímico del líquido de ascitis: trasudado, con un gradiente sero-ascítico de albúmina (GASA) de 1,78. Estudio bacteriológico del líquido de ascitis sin desarrollo.
Ecografía abdominal: ascitis severa, hepatomegalia irregular con ecogenicidad aumentada y esplenomegalia homogénea. Estudio Doppler del sistema portal: obstrucción de la vena suprahepática media y estrechamiento de la vena suprahepática derecha, con flujo de baja velocidad y escasamente fásico, invertido en algunas ramas; vena suprahepática izquierda y vena porta permeables y con flujo hepatófugo; VCI permeable y con flujo fásico.
Dado el antecedente de anafilaxia a medios de contraste en la paciente, no se realizó estudio imagenológico vascular contrastado.
Fibroesofagogastroduodenoscopía: várices esofágicas grado III con cherry red spots y antritis aguda.
Se planteó ascitis secundaria a hipertensión portal en función de estos hallazgos. Con respecto a la etiología de la hipertensión portal, se planteó el SBC dadas las alteraciones vasculares evidenciadas por ecografía Doppler.
Se inició tratamiento con diuréticos y betabloqueantes. También se realizó banding de várices esofágicas. En la evolución, la paciente presentó hemorragia digestiva masiva vinculada a la caída de la escara; ingresó a unidad de cuidados intensivos y se colocó una sonda de Sengstaken-Blakemore. Se realizó reposición sanguínea, administración de terlipresina y profilaxis de la encefalopatía hepática.
Posteriormente se realizó la búsqueda de estados protrombóticos para determinar la etiología del SBC: antitrombina III, proteínas C y S con resultados normales; el factor V Leiden y la mutación del gen de la protrombina (G20210A) fueron negativos; la mutación Jak 2 fue negativa y se descartó la hemoglobinuria paroxística nocturna mediante citometría de flujo; los anticuerpos IgM anti-beta 2 glicoproteína I fueron positivos (> a 100 U), resultado confirmado a las 12 semanas con una segunda determinación; los anticuerpos anticardiolipina y anticoagulante lúpico fueron negativos. Con estos resultados se diagnosticó trombofilia adquirida: SAF.
Seguidamente se inició tratamiento anticoagulante con heparinas de bajo peso molecular y superposición con warfarina. La paciente evolucionó refractaria al tratamiento médico. El caso se evaluó en conjunto con la Unidad de Trasplante Hepático del Hospital Central de las Fuerzas Armadas. La colocación de una derivación portosistémica intrahepática transyugular (conocida en inglés con la sigla TIPS, Transjugular Intrahepatic Portosystemic Shunt) no representaba una buena opción terapéutica en función del cuadro clínico y la anatomía vascular de la paciente. Por esta razón, se decidió realizar un trasplante hepático. Como complicación posterior al trasplante presentó un rechazo inicial que requirió tratamiento con altas dosis de corticoesteroides. La paciente presentó luego una buena evolución clínica, realizando controles periódicos posteriores.
Discusión
El cuadro clínico del SBC es heterogéneo y depende de la extensión y rapidez de instalación de la obstrucción, así como de la presencia de circulación colateral como mecanismo compensador. El espectro de manifestaciones clínicas abarca desde formas asintomáticas hasta el fallo hepático fulminante1,3,4. Se describen tres formas de presentación clínica según el tiempo de evolución2):
- Aguda (20%).
- Subaguda (40%), con signos y síntomas menores a seis meses de evolución y sin evidencia de cirrosis.
- Crónica (40%), con signos y síntomas mayores a seis meses de evolución y con evidencia de hipertensión portal o cirrosis, o ambas.
Los síntomas incluyen dolor abdominal, ascitis, ictericia, hepatomegalia, esplenomegalia, edemas, encefalopatía y/o sangrado gastrointestinal. El diagnóstico surge como un hallazgo ecográfico en un grupo de pacientes, particularmente en aquellos con formas subagudas o crónicas.
Los estudios de imagen juegan un papel crucial en el diagnóstico temprano del SBC. El estudio inicial en el caso de la paciente analizada fue una ecografía abdominal con Doppler. Este estudio tiene una alta sensibilidad y especificidad, pero se deben cumplir dos condiciones fundamentales para obtener el mayor rendimiento diagnóstico: su realización por personal experimentado y el aporte del dato clínico con la sospecha diagnóstica5. La ausencia de flujo o la presencia de flujo retrógrado en las venas hepáticas o en la VCI apoyan el diagnóstico de SBC5. La tomografía computada y la resonancia nuclear magnética de abdomen son otras técnicas de imagen complementarias que pueden demostrar la oclusión de las venas hepáticas o de la VCI.
El diagnóstico de SBC debería ser considerado en cualquier paciente con enfermedad hepática aguda o crónica e hipertensión portal, especialmente cuando no existen otras causas evidentes5. En el caso analizado se excluyeron otras causas más frecuentes de enfermedad hepática crónica: viral, alcohólica, autoinmune, esteatohepatitis no alcohólica, hemocromatosis y enfermedad de Wilson.
El SBC puede clasificarse desde el punto de vista etiológico como primario, cuando la obstrucción al flujo hepático se debe a una lesión intraluminal (por ejemplo, trombosis), o secundario, cuando la causa de la obstrucción vascular es una compresión extrínseca debida a tumores, abscesos o quistes1,6. En el 80% de los pacientes con SBC primario puede identificarse un trastorno subyacente. El 20% de casos restantes son catalogados como idiopáticos7. La etiología del SBC primario es a menudo multifactorial. Se observan uno o más estados protrombóticos en al menos el 75% de los casos1,3.
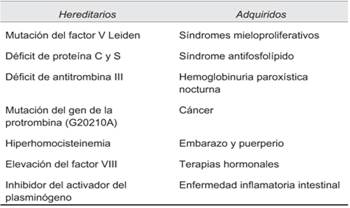
Con respecto a los estados protrombóticos, estos se clasifican según su origen en adquiridos y hereditarios. Esta clasificación se describe en la (tabla 2). En Occidente, las causas adquiridas más frecuentes son las enfermedades hematológicas, principalmente los síndromes mieloproliferativos crónicos (policitemia vera, trombocitemia esencial, metaplasia mieloide agnogénica). Otras causas adquiridas incluyen la hemoglobinuria paroxística nocturna, el síndrome antifosfolipídico, la enfermedad inflamatoria intestinal, las vasculitis, las enfermedades autoinmunes sistémicas, las neoplasias sólidas, el uso de anticonceptivos orales y el embarazo7. Las causas hereditarias descritas son la mutación del factor V Leiden, la mutación del gen de la protrombina (G20210A), el déficit de proteínas C, S y antitrombina III, entre otras.
El diagnóstico etiológico definitivo en el caso analizado fue SAF. Este síndrome, descrito por primera vez por Hughes en el año 1983, se caracteriza por la presencia de trombosis tanto arteriales como venosas, abortos de repetición o muerte fetal, entre otras manifestaciones, junto a la presencia de anticuerpos antifosfolipídicos (AAF)8,9. El SAF presenta naturaleza autoinmune y su diagnóstico debe realizarse en base a una serie de criterios clínicos e inmunológicos; estos criterios se exponen en la (tabla 3). Es importante recordar que el SAF puede clasificarse como primario o idiopático, o asociado a otra enfermedad autoinmune, con mayor frecuencia lupus eritematoso sistémico, teniendo implicancia en el estudio y seguimiento evolutivo de estos pacientes.

La asociación de SAF con SBC presenta escasos reportes en la literatura, fundamentalmente derivada de reportes de casos aislados. Espinosa y colaboradores describieron las características clínicas e inmunológicas de 43 pacientes con SBC y SAF; 32 pacientes presentaron SAF primario, mientras que los 11 pacientes restantes presentaron SAF secundario (8 de ellos debidos a lupus eritematoso sistémico)10. En este estudio, el SBC constituyó la manifestación inicial de SAF en 28 pacientes (65%), tal como sucedió en el caso analizado. Desde el punto de vista inmunológico, el 77 % de los pacientes presentó inhibidor lúpico positivo, mientras que 94% presentó anticuerpos anticardiolipinas positivos. En otros trabajos más recientes se ha reportado también la positividad para anticuerpos anti-beta 2 glicoproteína I en pacientes con SBC asociado a SAF11-13.
Cabe destacar que los anticuerpos anti-beta 2 glicoproteína I se consideran más específicos pero menos sensibles para el diagnóstico de SAF que los anticuerpos anticardiolipinas14. Aunque la positividad para los anticuerpos anti-beta 2 glicoproteína I de isotipo IgG e IgM se incluye en los criterios de clasificación de SAF, los trabajos realizados en relación con su significancia clínica han arrojado datos inconsistentes. En un estudio que incluyó 64 pacientes con SAF primario con solo un AAF detectado, más del 50% de los pacientes fueron positivos para anti-2 glicoproteína I del isotipo IgM15. Por el contrario, otros estudios han reportado una asociación mayor entre anticuerpos anti-beta 2 glicoproteína I del isotipo IgG y manifestaciones clínicas de SAF comparado con anticuerpos anti-beta 2 glicoproteína I del isotipo IgM, e incluso sugieren que estos últimos se asocian con menor probabilidad a SAF que otros isotipos16. Sin embargo, un resultado persistentemente positivo de anticuerpos anti-beta 2 glicoproteína I del isotipo IgM sería útil para establecer el diagnóstico de SAF en pacientes individualizados que se presenten con complicaciones trombóticas u obstétricas, como fue el caso de esta paciente16. De este modo, el planteo diagnóstico de SAF se sustentó en el resultado positivo en dos determinaciones de anticuerpos anti-beta 2 glicoproteína I del isotipo IgM separadas en el tiempo (que constituye a la fecha criterio diagnóstico de SAF), y en la ausencia de otras causas de trombofilia demostrables que pudieran relacionarse con el evento trombótico.
En los pacientes con SBC se recomienda seguir un algoritmo terapéutico “paso a paso”17. Todos los pacientes con diagnóstico de SBC deben recibir anticoagulación en forma precoz e indefinida para reducir el riesgo de extensión y prevenir la aparición de nuevos fenómenos trombóticos1,18. Esta terapia debe iniciarse con heparinas de bajo peso molecular por al menos cinco a siete días, administrando concomitantemente warfarina (con un INR objetivo entre 2 y 3). La oportunidad de inicio de anticoagulación debe evaluarse cuidadosamente, sobre todo por el mayor riesgo en estos pacientes de desarrollar complicaciones hemorrágicas vinculadas a la hipertensión portal (tal y como sucedió en el caso analizado), lo que hace esencial una correcta profilaxis del sangrado variceal. Si se confirma un estado protrombótico que amerite un tratamiento específico, el mismo debe iniciarse en forma concomitante.
Cuando existen estenosis segmentarias o parciales (60% en VCI, 25%-30% en venas hepáticas), se puede realizar angioplastia para restablecer el drenaje fisiológico portal y sinusoidal18. El procedimiento es efectivo y relativamente sencillo, con la posibilidad de un abordaje transyugular o transhepático19. La reestenosis posprocedimiento es frecuente, pero puede reducirse con la colocación de un stent.
En los pacientes con mala respuesta al tratamiento médico y que no son pasibles de tratamiento con angioplastia o stenting, o ambos, hay que considerar el empleo de técnicas de derivación porto-sistémica. Una opción es la realización de shunts quirúrgicos, como la anastomosis porto-cava latero-lateral. Los shunts quirúrgicos son inefectivos si existe asociada trombosis o compresión importante de la VCI1. Otra opción terapéutica es la colocación de una derivación portosistémica intrahepática transyugular. Esta técnica es de elección debido a la menor morbimortalidad, su fácil colocación y al manejo sencillo de las estenosis tardías18. También se utiliza como terapia puente al trasplante hepático1.
Finalmente, el trasplante hepático es el tratamiento de elección cuando el resto de las medidas terapéuticas han fracasado3. Los pacientes con presentación fulminante o cirrosis establecida con deterioro severo de la función hepática también deben ser considerados candidatos para este tratamiento18. En esta paciente se decidió el trasplante hepático dada la imposibilidad de realizar otras medidas terapéuticas para la hipertensión portal.
Es importante destacar que el diagnóstico y tratamiento precoz de los pacientes con SBC impacta directamente en el pronóstico del paciente. La mortalidad de los pacientes con SBC sin tratamiento es elevada, con cifras alrededor de 90% a los tres años de realizado el diagnóstico 20. Sin embargo, las tasas de sobrevida de pacientes con SBC tratados son buenas, con alrededor de 75% de pacientes vivos a los tres años21. La muerte se produce en la mayoría de los casos por insuficiencia hepática progresiva.
Conclusiones
Las enfermedades vasculares del hígado se deben tener presentes como causa de enfermedad hepática, fundamentalmente en los pacientes con predominio de las manifestaciones de hipertensión portal. Se presentó un caso de SBC secundario a SAF, asociación infrecuente en la literatura. El pronóstico de estos pacientes ha mejorado con el abordaje multidisciplinario y el acceso a diversas modalidades terapéuticas. El tratamiento debe ser individualizado según la presentación clínica. El trasplante hepático se plantea como medida de salvataje cuando el resto de los tratamientos instaurados ha fracasado.

