










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkRevista Médica del Uruguay
versión On-line ISSN 1688-0390
Rev. Méd. Urug. vol.31 no.3 Montevideo set. 2015
Pneumocistis jiroveccii como causa de fiebre de origen desconocido en paciente con dermatomiositis y tromboembolismo pulmonar
Dres. Gonzalo Méndez*, Rodrigo Andrade†, Maynés López‡, Laura Llambí§
Resumen
La dermatomiositis (DM) al igual que otras enfermedades autoinmunes ha sido reportada en varios estudios como un factor de riesgo para enfermedad tromboembólica venosa (ETEV). A su vez, debido a las alteraciones en la inmunidad causadas por la propia enfermedad, sumado a la inmunodepresión propia del tratamiento, estos pacientes son propensos a complicaciones infecciosas, muchas de ellas por gérmenes oportunistas como el Pneumocistis jirovecci (PJ). Los criterios diagnósticos de fiebre de origen desconocido (FOD) han estado durante largo tiempo en debate. En los últimos años se han propuesto nuevas categorías distintas a la FOD clásica, como ser la que afecta a pacientes inmunodeprimidos, donde tanto las etiologías como las formas de presentación de las mismas varía. Se han descrito más de 200 causas de FOD dentro de las cuales se incluye PJ. Se presenta el caso clínico de una paciente a la cual se le realizó diagnóstico de DM y que pese a la tromboprofilaxis presentó un episodio de tromboembolismo pulmonar. A su vez, durante la internación presentó una FOD. Luego de una búsqueda exhaustiva se aisló PJ y se realizó tratamiento para el mismo con excelente respuesta clínica.
Palabras clave: PNEUMOCYSTIS JIROVECII FIEBRE DE ORIGEN DESCONOCIDO DERMATOMIOSITIS EMBOLIA PULMONAR
Key words: PNEUMOCYSTIS JIROVECII FEVER OF UNKNOWN ORIGIN DERMATOMYOSITIS PULMONARY EMBOLISM
* Ex Residente de Medicina Interna Clínica Médica A, Hospital de Clínicas Dr. Manuel Quintela, Facultad de Medicina, Universidad de la República. Uruguay.
† Asistente de Clínica Médica A, Hospital de Clínicas Dr. Manuel Quintela, Facultad de Medicina, Universidad de la República. Uruguay.
‡ Ex Asistente de Clínica Médica A, Hospital de Clínicas Dr. Manuel Quintela, Facultad de Medicina, Universidad de la República. Uruguay.
§ Profesora Agregada de Clínica Médica A, Hospital de Clínicas Dr. Manuel Quintela, Facultad de Medicina, Universidad de la República. Uruguay.
Correspondencia: Dr. Gonzalo Méndez. Clínica Médica A, Hospital de Clínicas Dr. Manuel Quintela. Montevideo, Uruguay.
Correo electrónico: jgonzalomendez82@gmail.com
Recibido: 25/5/15 Aprobado: 10/8/15
Caso clínico
Paciente de sexo femenino, de 55 años de edad, con antecedentes personales de ser extabaquista sin historia de enfermedad tabaco dependiente.
Ingresó a sala de medicina por debut de dermatomiositis (DM) y se inició tratamiento con prednisona (1 mg/kg/día), azatioprina (150 mg/día) y tromboprofilaxis.
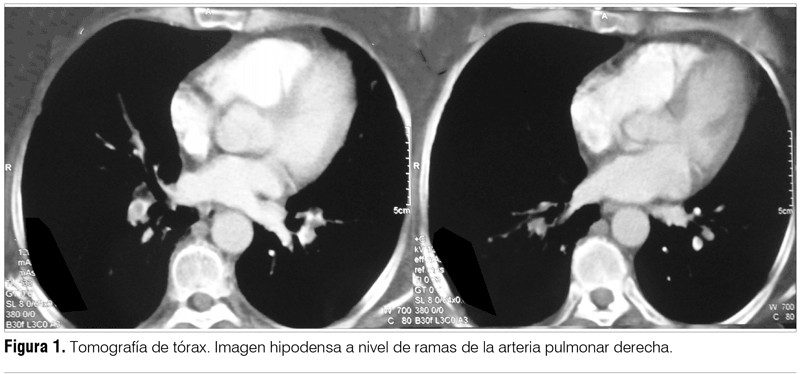
Establecido el diagnóstico de dicha enfermedad autoinmune, se realizó valoración en búsqueda de proceso neoplásico oculto sin evidencia del mismo. De la tomografía de tórax realizada por dicho motivo se destaca como hallazgo la presencia de un tromboembolismo pulmonar (TEP) (figura 1). Dado el hallazgo, se inició anticoagulación.
Presentó buena evolución, aunque persistió sintomática por varias semanas, permaneciendo internada para completar la valoración referida.
Durante la internación presentó registro febril de 38,5 °C axilar, sin foco clínico evidente ni evidencia de empuje de su enfermedad autoinmune. La fiebre persistió durante cuatro semanas con estabilidad clínica mantenida. Con diagnóstico de fiebre de origen desconocido (FOD) se plantearon las siguientes causas: infecciosa, neoplásica, autoinmune o vinculada a TEP.
Se solicitó valoración paraclínica: hemograma con leucopenia de 1.500/mm3 y linfopenia de 70/mm3, sin neutropenia. VES (velocidad de eritrosedimentación): 40 mm/h; PCR (proteína C reactiva): 90 mg/l; CPK (creatina-fosfocinasa): 200 UI/L. Radiografía de tórax y ecografía de abdomen y aparato urinario normal. Urocultivo y hemocultivo en dos oportunidades sin desarrollo. Ecocardiograma transtorácico sin evidencia de endocarditis infecciosa. Dado que se trataba de una FOD nosocomial y en una paciente inmunodeprimida se solicitó además una nueva tomografía de tórax, abdomen y pelvis sin cambios con la previa. Fibrobroncoscopía con lavado bronquioalveolar que no aisló germen. Estudio del líquido cefalorraquídeo normal. Fibrocolonoscopía normal.
PET SCAN evidenció a nivel de campos pulmonares lesiones hipermetabólicas compatibles con proceso inflamatorio, sin otros hallazgos.
Al mes de iniciada la fiebre comenzó con disnea de esfuerzo clase funcional III, tos seca, y al examen una polipnea de 24 rpm, MAV (murmullo alvéolo-vesicular) presente bilateralmente, sin estertores. Saturación de oxígeno de 95% que descendía al deambular a 88%.
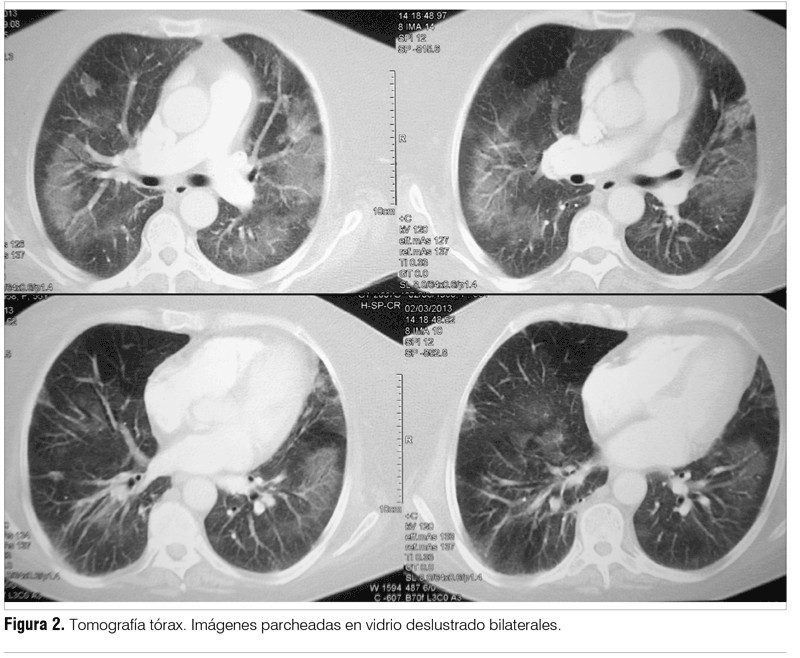
Dada la peoría clínica se inició tratamiento antibiótico empírico para cubrir gérmenes inespecíficos y neumocitosis con piperacilina, tazobactam y trimetoprim sulfametoxazol. Se solicitó nueva tomografía de tórax que evidenció un patrón parcheado en vidrio deslustrado de forma bilateral (figura 2) y una segunda fibrobroncoscopía con lavado bronquioalveolar, el resultado del lavado confirmó la presencia de Pneumocistis jiroveccii (PJ).
La paciente no reiteró registros febriles desde el inicio del antibiótico y presentó una franca mejoría en lo respiratorio. Permaneció con estabilidad en lo cutáneo-muscular y con INR (International Normalised Ratio) en rango se otorgó el alta a domicilio. Actualmente continúa con controles ambulatorios con buena evolución.
Discusión
Dermatomiositis y enfermedad tromboembólica venosa
La asociación de enfermedad tromboembólica venosa (ETEV) con enfermedades autoinmunes (EAI) ha sido ampliamente estudiada y reportada. Un trabajo realizado en Suecia con 535.538 pacientes internados con 33 EAI analizó el riesgo de TEP al año y en los siguientes diez años. Se encontró un riesgo aumentado en todas ellas, siendo el riesgo más alto en los pacientes con dermatomiositis/polimiositis (DM/PM) con 16,4 al año, reduciéndose luego a 3,4(1). Esta asociación de DM con ETEV fue corroborada también en otros estudios(2-4). La alta incidencia, así como el patrón temporal hallado, es atribuido a la etapa de miositis activa, así como a la movilidad reducida que lleva la propia enfermedad. En el caso presentado la ocurrencia de TEP, inicialmente asintomático, coincidió temporalmente con el diagnóstico de DM. Otros factores asociados con la ocurrencia de ETEV en pacientes con DM son la edad y el uso de inmunoglobulina(2-5).
Fiebre de origen desconocido y Pneumocistis jiroveccii
En 1961, se establecieron los criterios clásicos para definir la fiebre de origen desconocido como aquella que dura más de tres semanas con temperatura mayor a 38,3 °C en varias ocasiones, y en la cual no se llega a un diagnóstico luego de una semana de estudio en un hospital(6).
Algunos autores plantean la necesidad de adaptar esta definición a los avances en la práctica médica. En cuanto al criterio temporal proponen que sea de un plazo de tres días de estudio en el paciente hospitalizado o tres visitas de forma ambulatoria(7), mientras que otros autores proponen un criterio cualitativo(8-12), definiéndolo como un período luego del cual no se llega a un diagnóstico habiendo realizado un estudio apropiado(10). Estos cambios no han sido validados y la mayoría de los estudios se continúan realizando con la definición clásica. Un segundo cambio propuesto en la definición es la inclusión de nuevas categorías de pacientes, como ser los infectados por el virus de inmunodeficiencia humana (VIH), los neutropénicos o los pacientes hospitalizados (tabla 1)(7). Estos pacientes presentan no solo etiologías distintas sino que presentan una evolución más tórpida y por lo tanto requieren una conducta terapéutica más agresiva.
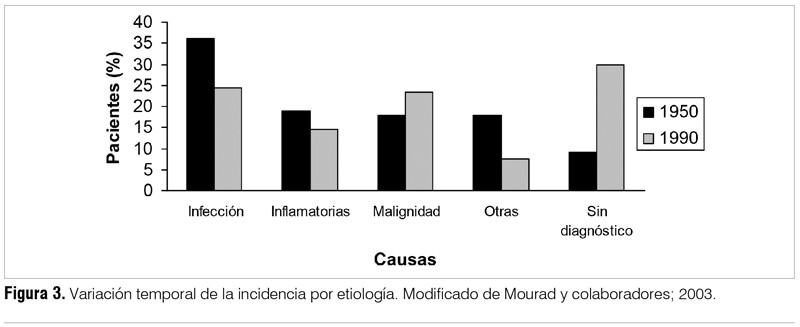
Se han identificado más de 200 causas de FOD y la lista continúa en aumento(13). Esto hace que no sea sencillo establecer un protocolo de estudio, que además debe estar adecuado a factores geográficos y económicos. Si bien no existen guías o recomendaciones basadas en la evidencia, algunos trabajos proponen estudios básicos que no estarían en discusión (tabla 2).(13). Se estima que el 2,9% de los pacientes internados presenta FOD(14), siendo las causas tradicionalmente agrupadas en cuatro categorías: infecciosas, neoplásicas, enfermedades inflamatorias e indeterminadas. En una revisión de 11 series con más de 1.000 pacientes entre 1952 y 1994, se reportó la incidencia de infecciones en 28%, enfermedades inflamatorias en 21%, neoplasias en 17% y 19% indeterminada. Cuando se analiza por décadas, se aprecia una disminución de las infecciones y de las neoplasias y un aumento de las causas no aclaradas (figura 3). Esto se plantea debido al diagnóstico más precoz de ciertas entidades, lo que determina que no cumplan el criterio temporal para ser consideradas FOD(15).
Dentro de las múltiples etiologías emergentes descritas como causa de FOD está el PJ(10,13). Si bien es una etiología rara, existen reportes de casos que la mencionan(16). PJ es una infección oportunista frecuente y bien conocida en el paciente portador de VIH, sin embargo su asociación con otras situaciones de inmunocompromiso es menos reconocida, aunque se encuentra descrita en la literatura. Los reportes de asociación de PJ en pacientes con EAI son raros antes de 1980, estando desde entonces en continuo aumento(17,18). Esto se explica por el uso generalizado de tratamientos inmunosupresores a altas dosis y combinados en estos pacientes, así como a la mejoría de las técnicas diagnósticas y al mayor conocimiento y búsqueda de la enfermedad(17).
En el caso de la DM/PM, Marie y colaboradores encontraron una incidencia de complicaciones infecciosas frecuentes (26%), siendo el 11,5% microorganismos oportunistas, entre las cuales estaba PJ. Esto, plantean, puede ser debido tanto a alteraciones de la inmunidad por la enfermedad en sí misma como por el tratamiento recibido(19). En el caso de la DM/PM, la incidencia de PJ estimada es de 27 casos/10.000 pacientes/año. Estos datos se obtuvieron de pacientes hospitalizados, lo cual sobreestima la incidencia(17).
El mayor factor de riesgo identificado para la infección por PJ en el no VIH es el uso de corticoides, uno de los tratamientos de primera línea para la DM/PM(20). En un estudio realizado con 116 pacientes, el 90,5% recibía corticoides. Si bien la media fue más alta, el 25% de ellos recibía solo 16 mg de prednisona al día y 25% lo presentaron a las ocho semanas, por lo que los autores marcan estos como puntos de corte para ser considerado factor de riesgo(21). Otros autores plantean el punto de corte en 20 mg de prednisona durante tres o cuatro semanas(22,23).
Otros factores de riesgo extra son la presencia de linfopenia y fibrosis pulmonar(24), la cual se encuentra presente en el 23,1% de los pacientes con DM/PM(25,26). El 91% de los pacientes en el momento de presentar PJ tenía una linfopenia menor a 1.500 y el 76% menor a 800 linfocitos(18).
Existen varias diferencias entre la infección por PJ en el portador de VIH y en el no VIH. En el no infectado por VIH tiene una presentación más abrupta(21), tanto en el inicio, con una duración media de síntomas hasta el diagnóstico más corto (5-6 días versus 28 días en el infectado por VIH)(18,27), así como mayor severidad de la misma con una tensión de oxígeno inferior(27,28). Los pacientes no infectados por VIH tienen menos microorganismos (lo cual vuelve el diagnóstico más dificultoso) y más neutrófilos, con una mayor respuesta inflamatoria(28).
La mortalidad en pacientes con PJ y EAI se estima entre 30% y 50%, la cual es más alta que en los pacientes infectados por VIH(17,18). De la alta incidencia y mortalidad de la infección, sumado a la alta eficacia de la quimioprofilaxis antibiótica, surge la recomendación de realizarla a todo paciente que presente factores de riesgo. La precaución es que en pacientes bajo tratamiento quimioterápico o con metrotexate puede aumentar la mielosupresión, así como en caso de presentar una complicación infecciosa el estar recibiendo un antibiótico disminuye la posibilidad de aislar el germen(29).
La profilaxis con trimetoprim sulfametoxazol se debería realizar en todo paciente con un desorden inmunológico de base y que reciba corticoides sistémicos de forma diaria y prolongada(29), así como suplementación con ácido folínico y seguimiento con hemogramas seriados(30).

Conclusión
El caso presentado alerta acerca de varias situaciones a tener en cuenta. Primero, la presentación de PJ como causa de FOD. Esto confirma que en general las causas de FOD no corresponden a patologías raras, sino a presentaciones atípicas de enfermedades bien conocidas. Segundo, ratifica la existencia de PJ en pacientes no infectados con VIH y dado que cada vez hay más pacientes en riesgo por el uso de corticoides o inmunosupresores, es necesario mantener un alto índice de sospecha para realizar un diagnóstico oportuno. Por último, recordar la importancia de la tromboprofilaxis y tener en cuenta el riesgo aumentado en los pacientes con EAI.
Abstract
Dermatomyositis (DM), the same as other auto-immune diseases, has been reported in several studies as a risk factor for a venous thromboembolic disease. Also, given immunity alterations caused by the disease itself, along with the immunodepression that characterizes treatment, these patients are likely to present infectious complications, many of them due to opportunistic germs such as Pneumocystis jirovecci (PJ).
Diagnostic criteria for fever of unknown origin (FUO) have been long discussed. Lately, new categories that are different to the classic FUO have been proposed, as is the one affecting immunodepressed patients, where both the etiology and presentation vary. Over 200 causes for FUO have been described, PJ being among them.
The clinical case of a female patient diagnosed with DM is described in the study, who, in spite of thromboprofilaxis presented a pulmonary thromboembolism episode. Likewise, during hospitalization, the patient evidenced FUO. After an extensive search, PJ was isolated and treatment was applied, resulting in an excellent clinical response.
Resumo
A dermatomiosite (DM), assim como outras patologias, tem sido descrita como um fator de risco para doença tromboembólica venosa (DTEV). As alterações da imunidade causadas poresta patologia, associadas à imunodepressão própria do tratamento fazem com que os pacientes sejam propensos a complicações infecciosas, muitas delas por microrganismos oportunistas como o Pneumocistis jirovecci (Pj).
Durante muito tempo os critérios diagnósticos de febre de origem indeterminada (FOI) foram causa de discussão. Nos últimos anos foram propostas novas categorias diferentes da FOI clássica, como por exemplo, a que afeta pacientes imunodeprimidos, nas quais, tanto as etiologias como as formas de apresentação variam. Mais de 200 causas de FOI, incluindo Pj, foram descritas.
Apresenta-se o caso clínico de uma paciente diagnosticada com DM e que, apesar da profilaxia tromboembólica, apresentou um episódio de tromboembolismo pulmonar. Durante a internação apresentou FOI; Pj foi isolado depois de uma exaustiva busca. A paciente recebeu o tratamento correspondente com excelente resposta clínica.
Bibliografía
1. Zöller B, Li X, Sundquist J, Sundquist K. Risk of pulmonary embolism in patients with autoimmune disorders: a nationwide follow-up study from Sweden. Lancet 2012; 379(9812):244–9.
2. Ramagopalan SV, Wotton CJ, Handel AE, Yeates D, Goldacre MJ. Risk of venous thromboembolism in people admitted to hospital with selected immune-mediated diseases: record-linkage study. BMC Med 2011;9:1.
3. Gaitonde SD, Ballou SP. Deep venous thrombosis in dermatomyositis. J Rheumatol 2008; 35(11):2288.
4. Selva-O’Callaghan A, Fernández-Luque A, Martínez-Gómez X, Labirua-Iturburu A, Vilardell-Tarrés M. Venous thromboembolism in patients with dermatomyositis and polymyositis. Clin Exp Rheumatol 2011; 29(5):846–9.
5. Zöller B, Li X, Sundquist J, Sundquist K. Autoimmune diseases and venous thromboembolism: a review of the literature. Am J Cardiovasc Dis 2012; 2(3):171–83.
6. Petersdorf RG, Beeson PB. Fever of unexplained origin: report on 100 cases. Medicine (Baltimore) 1961; 40:1–30.
7. Durack DT, Street AC. Fever of unknown origin-reexamined and redefined. Curr Clin Top Infect Dis 1991; 11:35–51.
8. De Kleijn EM, Vandenbroucke JP, van der Meer JW. Fever of unknown origin (FUO). I A. prospective multicenter study of 167 patients with FUO, using fixed epidemiologic entry criteria. The Netherlands FUO Study Group. Medicine (Baltimore) 1997; 76(6):392–400.
9. De Kleijn EM, van Lier HJ, van der Meer JW. Fever of unknown origin (FUO). II. Diagnostic procedures in a prospective multicenter study of 167 patients. The Netherlands FUO Study Group. Medicine (Baltimore) 1997; 76(6):401–14.
10. Knockaert DC, Vanderschueren S, Blockmans D. Fever of unknown origin in adults: 40 years on. J Intern Med 2003; 253(3):263–75.
11. Vanderschueren S, Knockaert D, Adriaenssens T, Demey W, Durnez A, Blockmans D, et al. From prolonged febrile illness to fever of unknown origin: the challenge continues. Arch Intern Med 2003; 163(9):1033–41.
12. Baicus C, Bolosiu HD, Tanasescu C, Baicus A. Fever of unknown origin-predictors of outcome. A prospective multicenter study on 164 patients. Eur J Intern Med 2003;14(4):249–54.
13. Arnow PM, Flaherty JP. Fever of unknown origin. Lancet 1997; 350(9077):575–80.
14. Iikuni Y, Okada J, Kondo H, Kashiwazaki S. Current fever of unknown origin 1982-1992. Intern Med 1994; 33(2):67–73.
15. Mourad O, Palda V, Detsky AS. A comprehensive evidence-based approach to fever of unknown origin. Arch Intern Med 2003; 163(5):545–51.
16. Jani K, Mehta NJ. Pneumocystis carinii pneumonia presenting as a fever of unknown origin in a patient without AIDS. Hear Lung J Acute Crit Care 2002; 31(1):50–2.
17. Ward MM, Donald F. Pneumocystis carinii pneumonia in patients with connective tissue diseases: the role of hospital experience in diagnosis and mortality. Arthritis Rheum 1999; 42(4):780–9.
18. Godeau B, Coutant-Perronne V, Le Thi Huong D, Guillevin L, Magadur G, De Bandt M, et al. Pneumocystis carinii pneumonia in the course of connective tissue disease: report of 34 cases. J Rheumatol 1994; 21(2):246–51.
19. Marie I, Hachulla E, Chérin P, Hellot M-F, Herson S, Levesque H, et al. Opportunistic infections in polymyositis and dermatomyositis. Arthritis Rheum 2005; 53(2):155–65.
20. Sepkowitz KA. Pneumocystis carinii pneumonia in patients without AIDS. Clin Infect Dis 1993; 17(Suppl 2):S416–22.
21. Yale SH, Limper AH. Pneumocystis carinii pneumonia in patients without acquired immunodeficiency syndrome: associated illness and prior corticosteroid therapy. Mayo Clin Proc 1996; 71(1):5–13.
22. Sepkowitz KA, Brown AE, Telzak EE, Gottlieb S, Armstrong D. Pneumocystis carinii pneumonia among patients without AIDS at a cancer hospital. JAMA 1992; 267(6):832–7.
23. Rodriguez M, Fishman JA. Prevention of infection due to Pneumocystis spp. in human immunodeficiency virus-negative immunocompromised patients. Clin Microbiol Rev 2004; 17(4):770–82.
24. Kadoya A, Okada J, Iikuni Y, Kondo H. Risk factors for Pneumocystis carinii pneumonia in patients with polymyositis/dermatomyositis or systemic lupus erythematosus. J Rheumatol 1996; 23(7):1186–8.
25. Maldonado F, Patel RR, Iyer VN, Yi ES, Ryu JH. Are respiratory complications common causes of death in inflammatory myopathies? An autopsy study. Respirology 2012; 17(3):455–60.
26. Marie I, Hachulla E, Chérin P, Dominique S, Hatron P-Y, Hellot M-F, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum 2002; 47(6):614–22.
27. Kovacs JA, Hiemenz JW, Macher AM, Stover D, Murray HW, Shelhamer J, et al. Pneumocystis carinii pneumonia: a comparison between patients with the acquired immunodeficiency syndrome and patients with other immunodeficiencies. Ann Intern Med 1984; 100(5):663–71.
28. Limper AH, Offord KP, Smith TF, Martin WJ. Pneumocystis carinii pneumonia. Differences in lung parasite number and inflammation in patients with and without AIDS. Am Rev Respir Dis 1989; 140(5):1204–9.
29. Sepkowitz KA. Pneumocystis carinii pneumonia without acquired immunodeficiency syndrome: who should receive prophylaxis? Mayo Clin Proc 1996; 71(1):102–3.
30. Thomas CF, Limper AH. Pneumocystis pneumonia. N Engl J Med 2004; 350(24):2487–98.


{kind=link}
{kind=link}
{kind=link}
{kind=link}