










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkRevista Médica del Uruguay
On-line version ISSN 1688-0390
Rev. Méd. Urug. vol.30 no.1 Montevideo Mar. 2014
Síndrome urémico hemolítico atípico:¿una entidad subdiagnosticada? Características clínicas y analíticas a propósito de dos casos
Dres. Ricardo Silvariño*, Gerardo Pérez†, Orlando Canzani‡, Patricia Larre Borges†, Nelson Acosta†, Óscar Noboa†
* Centro de Nefrología, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Departamento de Urgencia y Medicina Interna, Sanatorio Americano, Federación Médica del Interior, Uruguay.
† Centro de Nefrología, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay.
‡ Cooperativa Médica de Florida (COMEF). Uruguay.
Correspondencia: Dr. Óscar Noboa, Hospital de Clínicas, Av. Italia s/n y Las Heras, piso 14, CP 11400, Montevideo, Uruguay.
Correo electrónico: onoboa@hc.edu.uy
Recibido: 28/11/13 Aceptado:31/3/14
Resumen
La microangiopatía trombótica (MAT) es una condición anátomo-patológica caracterizada por lesión de la pared vascular con engrosamiento parietal, edema y desprendimiento de células endoteliales de la membrana basal. Clínicamente se presenta como anemia hemolítica microangiopática, oclusión microvascular por trombos plaquetarios y trombocitopenia. Puede ser consecuencia de una alteración primaria en el complejo sistema que regula la relación entre endotelio y coagulación u ocurrir en el contexto de una alteración sistémica. Dos fenotipos de la enfermedad son el púrpura trombocitopénico trombótico (PTT) y el síndrome urémico hemolítico (SUH). La disponibilidad de herramientas terapéuticas hace necesario un rápido reconocimiento de este mecanismo patogénico para poner en marcha una terapéutica precoz. Se presentan dos casos de SUH atípico y a partir de ellos se revisan los principales aspectos diagnósticos, terapéuticos y pronósticos de la enfermedad.
Palabras clave: SÍNDROME HEMOLÍTICO-URÉMICO
MICROANGIOPATÍAS TROMBÓTICAS
INTERCAMBIO PLASMÁTICO
Key words: HEMOLYTIC-UREMIC SYNDROME
THROMBOTIC MICROANGIOPATHIES
PLASMA EXCHANGE
Introducción
La microangiopatía trombótica (MAT) es una condición anátomo-patológica caracterizada por la lesión de la pared vascular (principalmente arteriolas o capilares) con engrosamiento parietal, edema y desprendimiento de células endoteliales de la membrana basal. Clínicamente se caracteriza por la presencia de anemia hemolítica microangiopática, oclusión microvascular por trombos plaquetarios y trombocitopenia, constituyendo esta la tríada clásica de la MAT(1-4). La MAT puede ser consecuencia de una alteración primaria en el complejo sistema que regula la relación entre endotelio y coagulación u ocurrir en el contexto de una alteración sistémica como se muestra en la tabla 1. Han sido descritas dos manifestaciones de la enfermedad patológicamente indistinguibles pero clínicamente diferentes: el síndrome urémico hemolítico (SUH) y el púrpura trombocitopénico trombótico (PTT). Dada la dificultad en identificar una u otra entidad muchos autores sugieren el uso del término integral PTT-SUH(1,4-7). El PTT-SUH comprende tradicionalmente dos fenotipos clínicos. La forma más frecuente (90% de los casos) se denomina PTT-SUH clásico o típico y se asocia usualmente con diarrea provocada por infección por Escherichia coli productora de la toxina Shiga (STEC)(2,8). Este microorganismo es capaz de unirse a receptores Gb3 de las células endoteliales y provocar la destrucción de estas de forma directa o a través de la activación de mecanismos inflamatorios y pro coagulantes. La mayoría de los pacientes con PTT-SHU-típico evolucionan satisfactoriamente al cabo de dos a tres semanas, pero 10% evoluciona hacia enfermedad renal crónica y 25% desarrolla secuelas renales permanentes(9). Un brote de esta entidad ocurrió en Alemania en 2011(10) con afectación registrada de 2.583 individuos de los cuales 855 cumplían con criterio de SUH, predominante adultos y mujeres(11). Dentro de los que fueron calificados como SUH, 91% cursó con injuria renal aguda y 60% tenía expresión neurológica(12). El otro fenotipo de enfermedad (PTT-SUH-atípico) afecta al 10% de los casos restantes. Se trata de un conjunto de enfermedades esporádicas, raras, no asociadas con diarrea y de peor pronóstico. Se presentan con compromiso neurológico o renal variable, o ambos. La mayoría de los pacientes presentan recurrencias y en los casos en que el riñón se compromete, más de 50% desarrolla insuficiencia renal terminal (IRT). Numerosos estudios han objetivado que el PTT-SHU-atípico posee un claro componente genético que se asocia con un déficit congénito o adquirido de ADAMST13 (enzima que cliva el factor de Von Willebrand regulando la agregación plaquetaria) o con mutaciones y polimorfismos en genes que codifican proteínas del sistema del complemento(13-20). Recientemente se ha implicado el desarrollo de PTT-SUH-atípico a mutaciones en el gen que codifica la diacilglicerolcinasa E, enzima vinculada a la regulación en menos de la activación plaquetaria(21). La disponibilidad de una terapéutica potencialmente curativa ha generado urgencia en realizar un correcto diagnóstico, y, por lo tanto, el rigor de los criterios de diagnóstico ha disminuido. En la actualidad solo la díada de la trombocitopenia y anemia hemolítica microangiopática es necesaria para establecer un diagnóstico de MAT e iniciar el tratamiento(1,3,4,7). Se presentan dos casos de SUH-atípico, se discuten aspectos diagnósticos y se actualiza la evidencia en referencia a las opciones terapéuticas actuales.

Caso 1
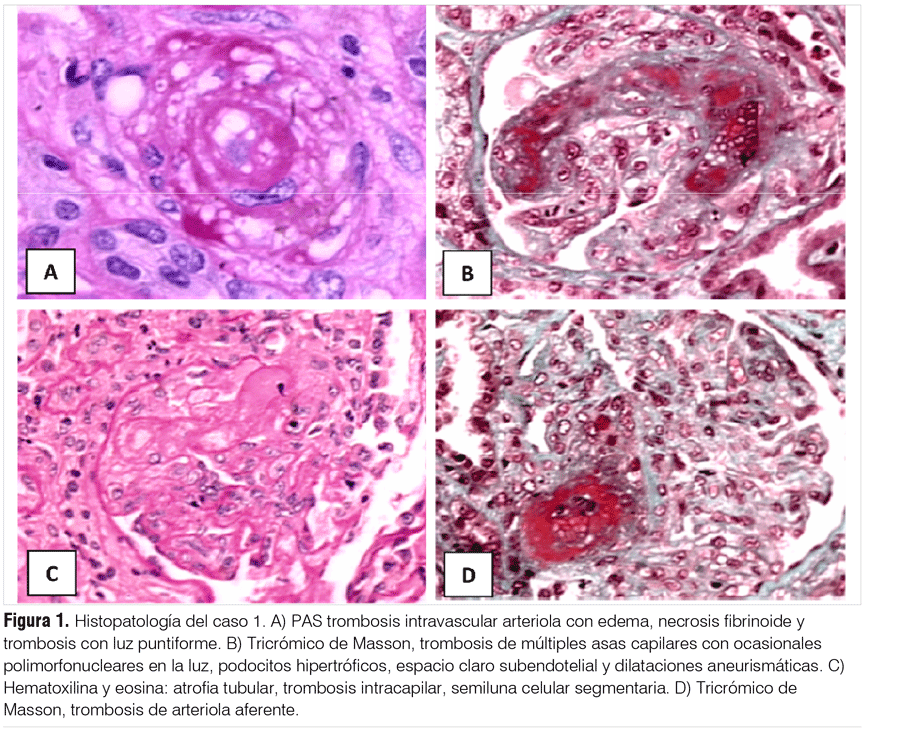
Paciente de sexo femenino, de 18 años edad. Historia de 15 días de evolución caracterizada por astenia y adinamia, agregando náuseas, vómitos y epigastralgia en los días previos a la consulta. Gingivorragia leve sin otros sangrados mucosos ni viscerales, con palidez cutáneo-mucosa progresiva. Disminución progresiva de la diuresis que no cuantificó. Negaba síntomas del tracto urinario superior e inferior, negaba diarrea. Cursó en apirexia. Como antecedentes personales relató anginas reiteradas en la infancia. Había iniciado el consumo de anticonceptivos orales desde el año previo a la consulta. No presentaba antecedentes familiares relevantes. Del examen clínico al ingreso destacaba buen estado general, anemia clínica, sin ictericia ni lesiones hemorragíparas asociadas. Taquicardia regular de 100 cpm, sin soplos ni roce pericárdico con presión arterial de 140/90 mmHg. Sin estertores. Se encontraba en oligoanuria. De la analítica de laboratorio (tabla 2) destacaban valores de insuficiencia renal severa, con hiperpotasemia y acidosis metabólica. La ecografía de aparato urinario objetivó la presencia de ambos riñones con tamaño y topografía habituales, sin litiasis, dilatación de cavidades, ni procesos expansivos. Se indica hemodiálisis y se realiza punción biópsica renal. El informe histológico destaca la presencia de 12 glomérulos, en tres se observa plegamiento de las basales con colapso de las luces capilares y aumento del espacio urinario. Los restantes presentaban trombosis de múltiples asas capilares con ocasionales polimorfonucleares en la luz, podocitos hipertróficos, espacio claro subendotelial y dilataciones aneurismáticas. Dos glomérulos agregaban a las lesiones descritas semilunas fibrocelulares segmentarias. Se visualizaban arteriolas con edema, necrosis fibrinoide y trombosis con luz puntiforme. En la inmunofluorescencia se observaban depósitos intracapilares con forma de trombos, positivo intenso para IgM, moderado para IgA, fibrinógeno e IgG. No se observaron depósitos de C3 ni de C1q (figura 1).
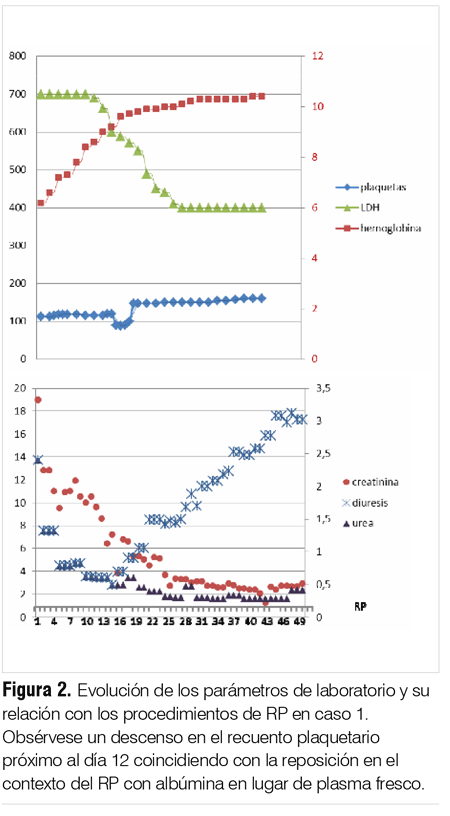
Los hallazgos histológicos asociados a trombocitopenia progresiva, anemia hemolítica microvascular e insuficiencia renal severa llevó al planteo de MAT a forma de PTT-SUH-atípico, por lo que se indicó recambio plasmático (RP), manteniéndose el soporte con hemodiálisis al inicio en forma diaria y luego en días alternos. Se realizaron un total de 17 procedimientos de RP con reposición con plasma fresco. En tres oportunidades la reposición se realizó con albúmina objetivándose un descenso en el recuento plaquetario por lo que se retornó a la reposición con plasma. La evolución fue a la mejoría clínica con recuperación progresiva de la diuresis, normalización del recuento plaquetario y descenso en los valores de urea y creatinina sin necesidad de tratamiento dialítico permanente. Al momento del alta presentaba una diuresis de 2.500 ml/día, urea 90 mg/dl, creatinina 2,4 mg/dl, bicarbonato venoso de 21 mmol/L, K 4,7 mEq/L, hemoglobina 10,5 g/L, plaquetas 177.000/mm3 y LDH 153 mg/dL. En la figura 2 se grafica la evolución de los parámetros de laboratorio y su relación con los procedimientos de RP.
Caso 2
Paciente de 22 años, de sexo femenino. Consultó por cuadro de 48 horas de evolución caracterizado por dolor abdominal, náuseas y vómitos en el contexto de infección respiratoria alta con fiebre de 38 ºC axilar. Niega ingesta de tóxicos o fármacos. Anuria durante las últimas 16 horas. Entre sus antecedentes se destacaba: cuatro años antes de este episodio, un cuadro agudo febril vinculado a infección respiratoria, caracterizado por afectación multisistémica, insuficiencia renal aguda anúrica con requerimiento de hemodiálisis de urgencia, trombocitopenia marcada y hemólisis asociados. Se realizó hemodiálisis durante tres semanas con recuperación progresiva del filtrado glomerular así como del resto de las disfunciones. En esa oportunidad, al presentar una recuperación lenta de la función renal, se realizó biopsia renal que mostró exclusivamente necrosis tubular aguda. Del examen clínico actual destacaba la presencia de anemia clínica, con ictericia leve de mucosas. La exploración linfoganglionar fue normal. En lo cardiovascular presentaba un ritmo regular de 76 pm sin soplos, con pulsos periféricos presentes y simétricos. El examen pleuropulmonar fue normal. A nivel abdominal, dolor difuso, sin signos de irritación peritoneal, sin hepatomegalia ni esplenomegalia. De la analítica de laboratorio (tabla 2) destacaban valores de insuficiencia renal severa, con hiperpotasemia y acidosis metabólica. La ecografía de aparato urinario objetivó la presencia de ambos riñones con tamaño y topografía habituales, sin litiasis, dilatación de cavidades, ni procesos expansivos. Se realizó hemodiálisis de urgencia. La asociación de anemia hemolítica microvascular, trombocitopenia e insuficiencia renal severa llevó al planteo de MAT a forma de PTT-SUH-atípico, por lo que se indicó RP, manteniéndose el soporte con hemodiálisis al inicio en forma diaria y espaciándose luego en el tiempo hasta suspenderse. Se realizaron un total de cinco procedimientos de RP con reposición con plasma fresco. La evolución fue a la mejoría clínica con recuperación progresiva de la diuresis, normalización del recuento plaquetario y descenso en los valores de urea y creatinina sin necesidad de tratamiento dialítico permanente. Al momento del alta presentaba una diuresis de 2.400 ml/día, urea 39 mg/dl, creatinina 1,1 mg/dl, bicarbonato venoso de 23 mmol/L, K 4,7 mEq/L, hemoglobina 13,5 g/L, plaquetas 241.000/mm3 y LDH 137 mg/dL.
Discusión
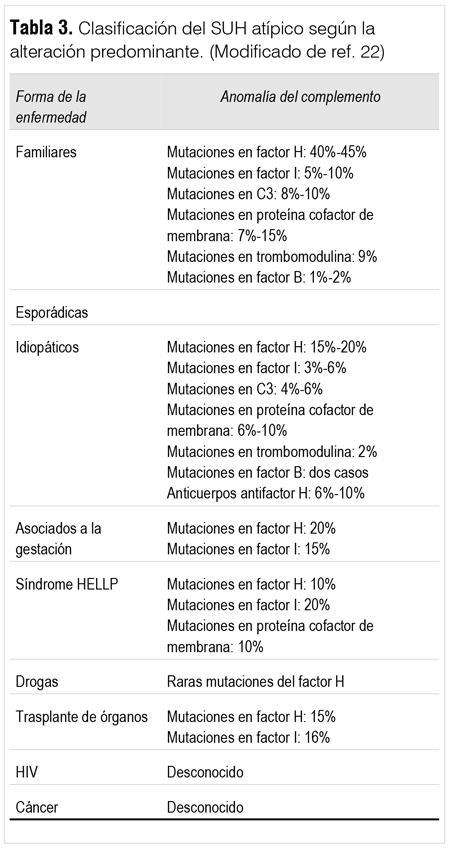
Presentamos dos casos de MAT a forma de PTT-SUH-atípico. A diferencia del síndrome urémico hemolítico clásico (asociado a infección por Escherichia coli productora de toxina Shiga y responsable del 90% de los casos de SUH) el PTT-SUH-atípico es una condición poco frecuente, reportada en 0,1 a 2 casos por millón de habitantes/año según diferentes series(22-26). Usualmente no se asocia a diarrea infecciosa, tiene un curso temporal recurrente con episodios vinculados a desencadenantes infecciosos u hormonales, presenta una mortalidad próxima a 25%, y en más de la mitad de los casos determina la necesidad de tratamiento sustitutivo de la función renal permanente(26,27). La presentación clínica polimorfa de los diferentes fenotipos de MAT ha llevado a establecer una clasificación basada en el mecanismo patogénico predominante en cada escenario (tabla 2)(26). El PTT-SUH-atípico es consecuencia de un déficit de ADAMTS13 congénito o adquirido (por anticuerpos anti-ADAMTS13) o a alteraciones congénitas o adquiridas del complemento, estando involucrados en su patogenia cuatro proteínas reguladoras de la vía alternativa (factor H, factor I, proteína cofactor de membrana y trombomodulina) y dos proteínas reguladoras de C3-convertasa (factor B y C3)(26). En la tabla 3 se muestran las alteraciones predominantes del complemento en cada escenario clínico(28). En nuestro medio no contamos con la capacidad de evaluar de forma sistemática las alteraciones del complemento responsables del PTT-SUH-atípico, por lo que el diagnóstico de la alteración molecular subyacente no fue realizado en los casos analizados. Aun con todas las herramientas disponibles en centros de referencia, hasta 40% de los casos quedan sin diagnóstico etiológico(28). El déficit absoluto o relativo de los factores reguladores del complemento explica el beneficio de la reposición con plasma fresco, ya que en este se encuentran presentes la totalidad de los factores mencionados. También se beneficia del RP el déficit de ADAMTS13, ya sea porque remueve los anticuerpos o porque restituye los niveles de ADAMTS13 con la reposición. En el primer caso analizado objetivamos un descenso del recuento plaquetario en el período en que la reposición fue hecha en base a albúmina. Este es otro elemento indirecto que aboga a que el déficit de factores reguladores subyace en la patogenia del PTT-SUH-atípico. En referencia a los desencadenantes posibles, múltiples autores insisten en la necesidad de un disparador de la lesión endotelial primaria, escenario sobre el que la disregulación del complemento determina la agregación plaquetaria y oclusión microvascular con las repercusiones clínicas mencionadas(26). En los casos presentados destacaba en el primero la ingesta durante el último año de anticonceptivos orales, y en el segundo caso el antecedente de infección respiratoria alta previo a cada episodio de insuficiencia renal severa. Dado que se cuenta con medidas terapéuticas eficaces, múltiples autores insisten en la necesidad de iniciar tratamiento frente a la presencia de trombocitopenia (plaquetas ? 100.000/mm3) asociada a anemia hemolítica microvascular (caída de la hemoglobina, aumento de bilirrubinas y/o LDH y presencia de esquistocitos en la lámina periférica)(5). Esta conducta se sustenta en el hecho de que la aparición de la péntada clásica (trombocitopenia, anemia hemolítica, fiebre, insuficiencia renal y alteraciones neurológicas) es expresión de enfermedad avanzada, a menudo con daño irreversible ya establecido y con alta mortalidad(29). En ambos casos el tratamiento fue relativamente precoz, esto puede explicar la reversibilidad de la insuficiencia renal severa en ambas circunstancias. El RP se considera el tratamiento de elección(30,31). Previo al uso del mismo la mortalidad del PTT-SUH era próxima a 90%, descendiendo a 20% luego del uso extendido de esta técnica(30). Debe iniciarse precozmente frente a la sospecha de PTT-SUH-atípico. Dado que el beneficio supera ampliamente el riesgo, algunos autores consideran que frente a la sospecha fundada de este trastorno debe iniciarse RP urgente, aunque de esta conducta se derive la sobreindicación del procedimiento(32-34). En el PTT-SUH-atípico, el RP es capaz de eliminar anticuerpos circulantes dirigidos contra los factores reguladores del complemento. La combinación del mismo con la reposición de factores aportados con el plasma fresco explica el efecto positivo de ambas técnicas combinadas(26). El uso de corticoides, si bien demostró cierto beneficio en referencia a la disminución de recaídas, en algunos grupos de pacientes con MAT(35) no ha sido específicamente evaluado en el SUH-atípico. El empleo de rituximab mostró algún beneficio en casos de PTT recurrente(36), sin embargo no se ha evaluado su utilidad en el contexto del SUH-atípico. El eculizumab es un nuevo anticuerpo monoclonal que actúa inhibiendo selectivamente la proteína del complemento C5, impide la formación de C5a y C5b e inhibe la formación del complejo de ataque de membrana(26). Fue aprobado en Estados Unidos por la Food and Drug Administration (FDA) en setiembre de 2011 para el tratamiento de SHU-atípico. Múltiples reportes de caso avalan su uso en este escenario clínico(26), pero lo elevado de su costo impide su utilización sistemática.
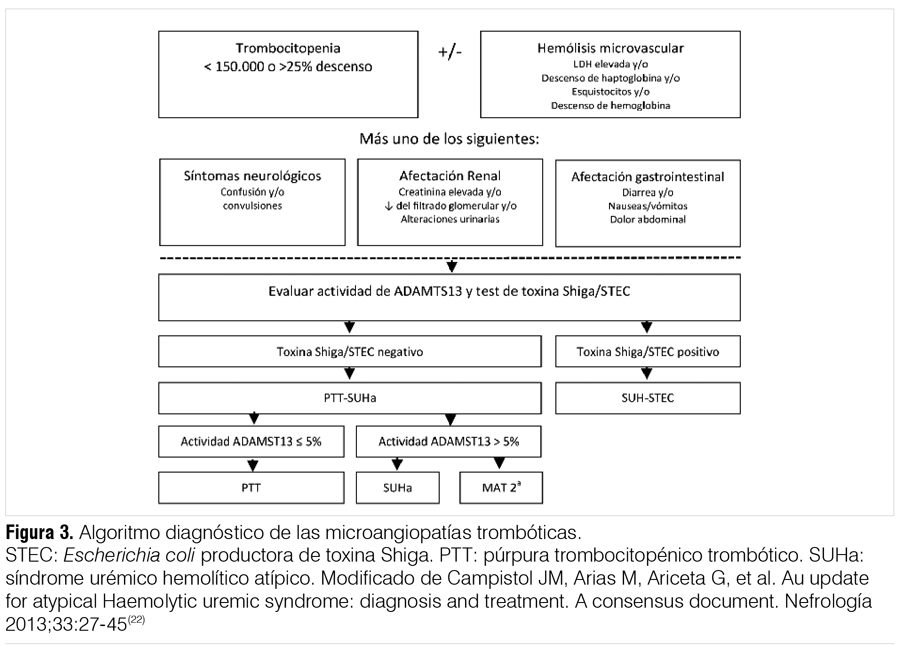
En la figura 3 se muestra un posible algoritmo diagnóstico frente a la sospecha de PTT-SUH-atípico.
Conclusiones
La MAT vinculada a PTT-SUH es una entidad muy poco frecuente. Puede acompañar múltiples situaciones patológicas y es un mecanismo patogénico que debe estar presente en el razonamiento clínico. De su diagnóstico precoz depende tener una conducta terapéutica apropiada. En los dos casos analizados destacan algunos aspectos muy relevantes. En el primero la biopsia renal nos obligó a reconocer aspectos clínicos que inicialmente no eran demasiado evidentes y condujo a la opción terapéutica, la misma fue realizada en el momento oportuno, con plaquetopenia leve. La respuesta al RPT y la reproducción de la plaquetopenia cuando la sustitución fue con albúmina permite reconocer que la reposición de plasma jugaba un rol fundamental, probablemente reponiendo un factor faltante en este caso. El segundo caso pone en evidencia aspectos clínicos relevantes como la eventualidad de que la biopsia renal no muestre más que una injuria renal aguda, que la patología puede tener un curso recurrente, puede ser desencadenada por infecciones, y resolverse espontáneamente. Aun en los centros de referencia que cuentan con herramientas diagnósticas para disecar la multiplicidad de mecanismos involucrados, el 40% de los casos queda sin diagnóstico de mecanismo patogénico, lo que hace más relevante contar con centros de referencia regionales que permitan una aproximación diagnóstica apropiada y una utilización prudente de los recursos terapéuticos.
Agradecimientos
A los Dres. Rosario Cuadro, Enrique Méndez, Pilar Varela, Alejandro Opertti y María García.
Abstract
Thrombotic microangiopathy (MAT) is an anatomo-pathological condition characterized by a lesion in the arterial wall with parietal thickening, edema and endothelial cells detached from the basement membrane. Clinically it is seen as a hemolytic microangiopathic anaemia, microvascular occlusion due to platelete count and thrombocytopenia. It could be a consequence of a primary alteration in the complex system that regulates the relationship between the endothelio and coagulation or occur within the context of a systemic alteration. Thrombotic thrombocytopenic purpura (PPT) and hemolytic uremic syndrome (HUS) are two phenotypes of the disease. Availability of therapeutic tools calls for the fast acknowledgement of this pathogenic mechanism to start up early therapeutic. The study presents two cases of atypical HUS and based on that reviews the main diagnostic, therapeutic and prognostic aspects.
Resumo
A microangiopatia trombótica (MAT) é uma entidade anatomopatológica caracterizada pela lesão da parede vascular com engrossamento parietal, edema e desprendimento de células endoteliais da membrana basal. Clinicamente se apresenta como anemia hemolítica microangiopática, oclusão microvascular por trombos plaquetários e trombocitopenia. Pode ser consequência de uma alteração primária no complexo sistema que regula a relação entre endotélio e coagulação ou no contexto de uma alteração sistêmica. Dois fenótipos da doença são a púrpura trombocitopênica trombótica (PTT) e a síndrome hemolítico-urêmica (SHU). A existência de recursos terapêuticos impõe o reconhecimento rápido deste mecanismo patogênico para começar o tratamento. Apresenta-se dois casos de SUH atípico e a faz-se a revisão dos principais aspectos diagnósticos, terapêuticos e prognósticos desta patologia.
Bibliografía
1. Ruggenenti P, Noris M, Remuzzi G. Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney Int 2001; 60(3):831-46.
2. George JN, Gilcher RO, Smith JW, Chandler L, Duvall D, Ellis C. Thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: diagnosis and management. J Clin Apher 1998; 13(3):120-5.
3. George JN, Vesely SK. Thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: diagnosis and treatment. Cleve Clin J Med 2001; 68(10):857-8, 860, 863-4.
4. George JN, Vesely SK, Terrell DR. The Oklahoma Thrombotic Thrombocytopenic Purpura-Hemolytic Uremic Syndrome (TTP-HUS) Registry: a community perspective of patients with clinically diagnosed TTP-HUS. Semin Hematol 2004; 41(1):60-7.
5. Rock GA, Shumak KH, Buskard NA, Blanchette VS, Kelton JG, Nair RC, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med 1991; 325(6):393-7.
6. George JN. How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood 2010; 116(20):4060-9.
7. George JN. Clinical practice: thrombotic thrombocytopenic purpura. N Engl J Med 2006; 354(18):1927-35.
8. Gadea M del P, Varela G, Bernadá M, Sirok A, Mota MI, Sabelli R, et al. Primer aislamiento en Uruguay de Escherichia coli productora de toxina Shiga del serotipo O157:H7 en una niña con síndrome urémico hemolítico. Rev Méd Urug 2004; 20(1):79–81.
9. Noris M, Remuzzi G. Hemolytic uremic syndrome. J Am Soc Nephrol 2005; 16(4):1035-50.
10. Frank C, Werber D, Cramer JP, Askar M, Faber M, an der Heiden M, et al; HUS Investigation Team. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N Engl J Med 2011; 365(19):1771-80.
11. Kielstein JT, Beutel G, Fleig S, Steinhoff J, Meyer TN, Hafer C, et al; Collaborators of the DGfN STEC-HUS registry. Best supportive care and therapeutic plasma exchange with or without eculizumab in Shiga-toxin-producing E. coli O104:H4 induced haemolytic-uraemic syndrome: an analysis of the German STEC-HUS registry. Nephrol Dial Transplant 2012; 27(10):3807-15.
12. Braune SA, Wichmann D, von Heinz MC, Nierhaus A, Becker H, Meyer TN, et al. Clinical features of critically ill patients with Shiga toxin-induced hemolytic uremic syndrome. Crit Care Med 2013; 41(7):1702-10.
13. Noris M, Mescia F, Remuzzi G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol 2012; 8(11):622-33.
14. Noris M, Remuzzi G. Genetics and genetic testing in hemolytic uremic syndrome/thrombotic thrombocytopenic purpura. Semin Nephrol 2010; 30(4):395-408.
15. Noris M, Remuzzi G. Complement factor h gene abnormalities in haemolytic uraemic syndrome: from point mutations to hybrid gene. PLoS Med 2006; 3(10):e432. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1626557/pdf/pmed.0030432.pdf. [Consulta: marzo 2014].
16. Noris M, Remuzzi G. Genetic abnormalities of complement regulators in hemolytic uremic syndrome: how do they affect patient management? Nat Clin Pract Nephrol 2005; 1(1):2-3.
17. Noris M, Remuzzi G. Are HUS and TTP genetically determined? Kidney Int 1998; 53(4):1085-6.
18. Noris M, Bresin E, Mele C, Remuzzi G. Atypical Hemolytic-Uremic Syndrome: 2007 Nov 16 [updated 2013 Aug 08]. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Smith RJH, et al, eds. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2014. Disponible en: http://www.ncbi.nlm.nih.gov/books/NBK1367/. [Consulta: marzo 2014].
19. Noris M, Ruggenenti P, Perna A, Orisio S, Caprioli J, Skerka C, et al. Hypocomplementemia discloses genetic predisposition to hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: role of factor H abnormalities. Italian Registry of Familial and Recurrent Hemolytic Uremic Syndrome/Thrombotic Thrombocytopenic Purpura. J Am Soc Nephrol 1999; 10(2):281-93.
20. Remuzzi G, Ruggenenti P, Colledan M, Gridelli B, Bertani A, Bettinaglio P, et al. Hemolytic uremic syndrome: a fatal outcome after kidney and liver transplantation performed to correct factor h gene mutation. Am J Transplant 2005; 5(5):1146-50.
21. Lemaire M, Frémeaux-Bacchi V, Schaefer F, Choi M, Tang WH, Le Quintrec M, et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet 2013; 45(5):531–6. Disponible en: http://www. ncbi. nlm.nih.gov/pmc/articles/PMC3719402/ pdf/nihms-474970. pdf. [Consulta: marzo 2014].
22. Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med 2009; 361(17):1676-87.
23. Constantinescu AR, Bitzan M, Weiss LS, Christen E, Kaplan BS, Cnaan A, et al. Non-enteropathic hemolytic uremic syndrome: causes and short-term course. Am J Kidney Dis 2004; 43(6):976-82.
24. Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, Carreras L, Aller Arranz E, Abarrategui Garrido C, et al. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci U S A 2007; 104(1):240–5. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1765442/ pdf/zpq240.pdf. [Consulta: marzo 2014].
25. Kavanagh D, Kemp EJ, Mayland E, Winney RJ, Duffield JS, Warwick G, et al. Mutations in complement factor I predispose to development of atypical hemolytic uremic syndrome. J Am Soc Nephrol 2005; 16(7):2150-5.
26. Campistol Plana JM, Arias M, Ariceta Iraola G, Blasco M, Espinosa M, Grinyó JM, et al. Actualización en síndrome hemolítico urémico atípico: diagnóstico y tratamiento. Documento de consenso. Nefrología 2013; 33(1):27–45. Disponible en: http://www.revistanefrologia.com/revistas/P1-E547/P1-E547-S3861-A11781.pdf. [Consulta: marzo 2014].
27. Cameron JS, Vick R. Letter: Plasma-C3 in haemolytic-uraemic syndrome and thrombotic thrombocytopenic purpura. Lancet 1973; 2(7835):975.
28. Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaimé F, Dragon-Durey MA, Ngo S, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol 2013; 8(4):554-62.
29. Wada H, Wakita Y, Nakase T, Shimura M, Hiyoyama K, Nagaya S, et al. Increased plasma-soluble fibrin monomer levels in patients with disseminated intravascular coagulation. Am J Hematol 1996; 51(4):255-60.
30. von Baeyer H. Plasmapheresis in thrombotic microangiopathy-associated syndromes: review of outcome data derived from clinical trials and open studies. Ther Apher 2002; 6(4):320-8.
31. Kremer Hovinga JA, Vesely SK, Terrell DR, Lämmle B, George JN. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood 2010; 115(8):1500-11.
32. Moake JL. Thrombotic microangiopathies. N Engl J Med 2002; 347(8):589-600.
33. Rizvi MA, Vesely SK, George JN, Chandler L, Duvall D, Smith JW, et al. Complications of plasma exchange in 71 consecutive patients treated for clinically suspected thrombotic thrombocytopenic purpura-hemolytic-uremic syndrome. Transfusion 2000; 40(8):896-901.
34. Nguyen L, Terrell DR, Duvall D, Vesely SK, George JN. Complications of plasma exchange in patients treated for thrombotic thrombocytopenic purpura. IV. An additional study of 43 consecutive patients, 2005 to 2008. Transfusion 2009; 49(2):392-4.
35. Balduini CL, Gugliotta L, Luppi M, Laurenti L, Klersy C, Pieresca C, et al; Italian TTP Study Group. High versus standard dose methylprednisolone in the acute phase of idiopathic thrombotic thrombocytopenic purpura: a randomized study. Ann Hematol 2010; 89(6):591-6.
36. George JN, Woodson RD, Kiss JE, Kojouri K, Vesely SK. Rituximab therapy for thrombotic thrombocytopenic purpura: a proposed study of the Transfusion Medicine/Hemostasis Clinical Trials Network with a systematic review of rituximab therapy for immune-mediated disorders. J Clin Apher 2006; 21(1):49-56.


{kind=link}
{kind=link}