










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkRevista Médica del Uruguay
On-line version ISSN 1688-0390
Rev. Méd. Urug. vol.28 no.1 Montevideo Mar. 2012
Enfermedades priónicas en el ser humano en Uruguay: registro de los últimos 25 años
Sara Lewin*, Abayubá Perna†, Ronald Salamano‡, Carlos Ketzoian§, Daniel Salinas¶, María Mirta Rodríguez††, Graciela Mañana‡‡
Cátedra de Neurología, Secciones de Neuroinfectología y Neuroepidemiología, Instituto de Neurología, Facultad de Medicina, Universidad de la República. Uruguay
Resumen
Introducción: las enfermedades priónicas son enfermedades de carácter degenerativo del sistema nervioso central de curso progresivo y desenlace fatal que presentan largos períodos de incubación antes de manifestarse clínicamente siendo hoy de gran interés científico dado que responden a un modelo autorreplicante proteico sin intervención de ácidos nucleicos. Asimismo tienen un carácter simultáneo de aparición esporádico, hereditario e infectante. La aparición de la encefalopatía bovina espongiforme y su consecuencia en el ser humano, la variante de la enfermedad de Creutzfeldt-Jacob, subrayan la necesidad de un control epidemiológico estricto en la materia.
Objetivo: describir la realidad de estas enfermedades en Uruguay en el período que media entre 1984 y 2009 inclusive.
Material y método: se realizó una revisión descriptiva y retrospectiva de casos clínicos de enfermedades priónicas diagnosticadas en nuestro país.
Resultados: se lograron identificar 42 casos de enfermedad de Creutzfeldt-Jakob en Uruguay (8 formas hereditarias y 34 formas esporádicas). La tasa de incidencia estimada global fue de 0,7 casos por millón de habitantes por año, considerando las formas probables y definitivas.
Conclusiones: en este trabajo se han detectado exclusivamente casos esporádicos y familiares de Creutzfeldt-Jacob, no se han detectado casos vinculados a la variante relacionada con la encefalopatía espongiforme bovina, ni a otros tipos de enfermedades priónicas que afectan a los seres humanos.
Dadas las características de la enfermedad y la distribución de neurólogos en todo el país, es posible realizar un relevamiento y una vigilancia epidemiológica bastante estricta de estas enfermedades en nuestro medio.
Palabras clave: ENFERMEDADES POR PRION
SÍNDROME DE CREUTZFELDT-JAKOB
URUGUAY
Keywords: PRION DISEASES
CREUTZFELDT-JAKOB SYNDROME
URUGUAY
* Sección de Neuroinfectología, Instituto de Neurología, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay.
† Profesor Adjunto de Neurología. Sección de Neuroinfectología y Neuroepidemiología, Instituto de Neurología, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay.
‡ Profesor Titular de la Cátedra de Neurología, Sección de Neuroinfectología, Instituto de Neurología, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay.
§ Profesor Agregado de Métodos Cuantitativos, Sección de Neuroepidemiología, Instituto de Neurología, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay.
¶ Neurólogo. Uruguay.
†† Profesora Titular del Departamento de Genética, Sección de Neurogenética, Instituto de Neurología, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay.
‡‡ Profesora Agregada del Departamento de Neuropatología, Instituto de Neurología, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay.
Correspondencia: Dra. Sara Lewin. Humberto 1º 4096 Apart. 305, CP 11400, Montevideo, Uruguay.
Correo electrónico: saraschutz1@hotmail.com
Recibido: 7/11/11
Aceptado: 9/3/12
Conflicto de intereses: los autores del presente artículo declaran que no existe conflicto de intereses.
Introducción
Las enfermedades priónicas o encefalopatías espongiformes transmisibles son enfermedades de carácter degenerativo del sistema nervioso central (SNC) de curso progresivo y desenlace fatal que presentan largos períodos de incubación antes de manifestarse clínicamente. Durante este período de silencio clínico no es posible detectarlas con las pruebas biológicas que existen en el momento.
La degeneración del SNC implica cambios espongiformes (espongiforme por el aspecto que presentan en la anatomía patológica) que involucran la corteza cerebral, los ganglios basales y el cerebelo, con depósito de material amiloideo, constituido por la proteína priónica.
Su denominación apunta a señalar la acumulación de una proteína anómala como el agente causal de estas encefalopatías, el prion (PRoteinaceus InfectiOus ageNt), que tiene un poderoso efecto neurotóxico, no desencadenando respuesta inmune ni inflamatoria en el huésped(1).
Esta proteína anómala (PrPsc) reúne una serie de singularidades. La primera es que resulta ser una variación conformacional de una proteína normal (PrPc) que se encuentra en la membrana celular de neuronas y otros tipos de células, de acción por el momento incierta en la fisiología celular. Esta proteína que se encuentra codificada en un gen (PRNP) del brazo corto del cromosoma 20, es una glicoproteína que tiene una configuración alfa-hélice. El prion tiene la misma constitución aminoacídica (estructura primaria) que la proteína normal, pero adopta una estructura secundaria de tipo beta, lo que facilita el plegamiento y la acumulación(2).
Esta variación conformacional le otorga al prion la calidad de ser altamente insoluble y resistente a la digestión por proteasas(3).
Se presume que el contacto de estas dos formas (la alfa y la beta) facilitaría la transformación de la proteína normal en anormal. No habiéndose detectado por el momento la existencia de material nuclear (ácido desoxiribonucleico [ADN] o ácido ribonucleico [ARN]) en el proceso de síntesis del prion.
La segunda singularidad a destacar es que dicha proteína tiene un carácter infectante ya sea a través de sustancias contaminadas en contacto con el encéfalo (implantes de duramadre, trasplantes de córnea, electroencefalografía con electrodos profundos), tratamiento parenteral de sustitución con hormona de crecimiento y gonadotrofinas hipofisarias, originadas en extractos de hipófisis de cadáveres, o de la ingesta de productos cárnicos contaminados de ganado portador de la encefalopatía espongiforme bovina (EEB) y que producen la variante de la enfermedad de Creutzfeldt-Jakob (vCJ) (4).
La tercera de las singularidades alude al carácter hereditario de estas afecciones, de hecho existen mutaciones en el gen PRNP que producen estas enfermedades (5).
Las enfermedades priónicas no son exclusivas del hombre, diferentes especies las poseen (scrapie en las ovejas, EEB o mal de la vaca loca, enfermedad caquectizante del alce, encefalopatía transmisible del visón, encefalopatía espongiforme felina, encefalopatía espongiforme de animales en cautiverio), siendo altamente probable que algunas de estas enfermedades sean consecuencia de la contaminación interespecies(6).
Las enfermedades priónicas en el ser humano se clasifican de la siguiente manera:
A) Enfermedades priónicas adquiridas
- Kuru
- Enfermedad de Creutzfeldt-Jakob iatrogénica
- Variante de la enfermedad de Creutzfeldt-Jakob
B) Enfermedades priónicas esporádicas
- Enfermedad de Creutzfeldt-Jakob (ECJ) esporádica
C) Enfermedades priónicas hereditarias
- Enfermedad de Creutzfeldt-Jakob hereditaria
- Enfermedad de Gerstmann-Straüssler-Scheinker
- Insomnio fatal familiar y esporádico
La ECJ esporádica es la más frecuente, constituye 85% de los casos conocidos, tiene una incidencia regular en todo el globo terráqueo de 0,5-1,5 casos por millón por año, apareciendo sobre los 50-70 años. Se desconoce su origen.
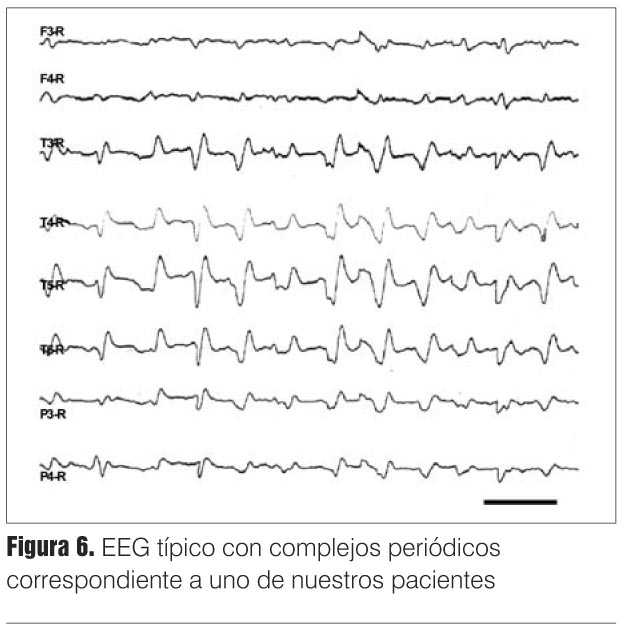
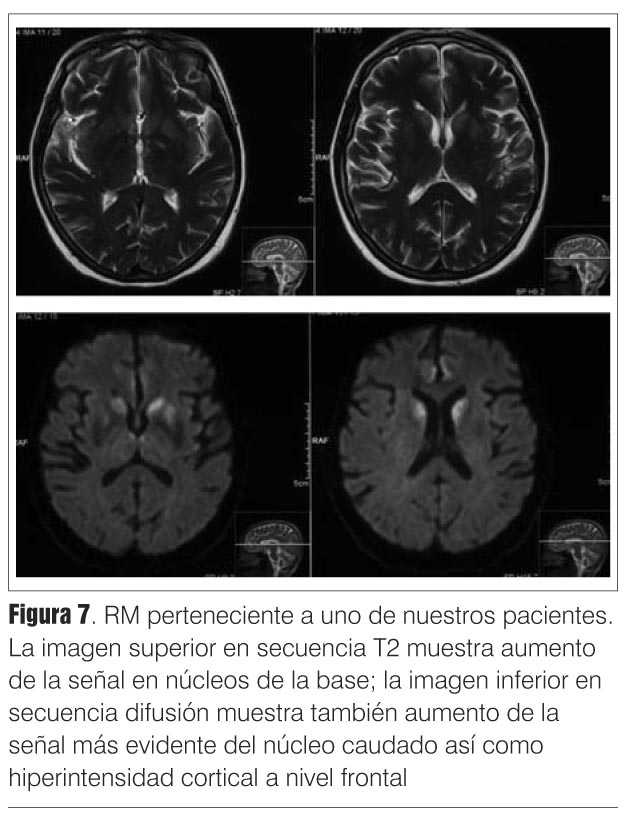
Se trata de una demencia mioclónica rápidamente progresiva que suma elementos piramidales, extrapiramidales parkinsonianos, tónico-frontales, cerebelosos, etcétera, y que lleva ineluctablemente a la muerte en cuestión de meses. El paciente puede presentar en algún momento de su evolución un electroencefalograma (EEG) típico de la afección (complejos periódicos trifásicos, de 0,5 - 2 c/seg) y un estudio de la proteína 14-3-3 en el líquido céfalo-raquídeo (LCR) (proteína de degradación neuronal) positivo. La resonancia magnética (RM) es de gran importancia en el diagnóstico siendo sus hallazgos bastante específicos; puede mostrar un aumento de señal en T2, secuencia FLAIR y especialmente difusión en la zona ganglio-basal así como en la corteza cerebral. Su sensibilidad ha sido comparada con la de la anatomía patológica. Sin embargo, no es todavía un criterio diagnóstico aceptado por la Organización Mundial de la Salud (OMS)(7-10).
La ECJ iatrógena fue descripta en el pasado en relación con contaminación de material en contacto con el cerebro tal como se describió y a la administración de hormona de crecimiento y gonadotropinas humanas como terapéutica de sustitución.
La ECJ hereditaria constituye el 13%-15% restante de las enfermedades priónicas en el ser humano; existen más de 25 mutaciones conocidas, todas ellas en el gen PRNP del brazo corto del cromosoma 20. Se trata de una herencia autosómica dominante, monogénica, de penetrancia incompleta. Habitualmente el debut de la enfermedad se hace en edades más tempranas y el curso de la encefalopatía es más prolongado(1,11).
La vCJ fue detectada en 1996, luego de la epidemia de EEB producida en el Reino Unido en los años precedentes. Se caracteriza por afectar a personas jóvenes, comienza con manifestaciones psiquiátricas que dan paso a síntomas sensitivos irritativos (parestesias, dolores), alteraciones autonómicas y luego demencia. El curso es más lento que la ECJ clásica. La RM muestra un aumento de señal en el núcleo pulvinar del tálamo en secuencia de T2, FLAIR y difusión. Desde el punto de vista anátomo-patológico conforma imágenes en la microscopía óptica que son denominadas "placas floridas"(11). A agosto de 2011 se han descripto 219 casos de vCJ (Reino Unido, Francia, Irlanda, Italia, Estados Unidos, Canadá, Arabia Saudita, Japón, Holanda, Portugal, España y Taiwán) (12).
La enfermedad de Gerstmann-Straüssler-Scheinker (GSS) es una enfermedad hereditaria que afecta en general a jóvenes. Predominan los síntomas y signos cerebelosos que luego dan lugar a un cuadro demencial. La enfermedad puede prolongarse por varios años. La anatomía patológica muestra acumulación de placas amiloideas en el cerebelo junto a la degeneración espongiforme(11).
El insomnio fatal familiar y esporádico es una entidad sumamente rara, caracterizada por dificultades progresivas en la conciliación del sueño, produciéndose un insomnio incoercible, agregándose luego síntomas sensitivos irritativos, alteraciones autonómicas y luego demencia(13,14).
Por último, el Kuru, enfermedad exclusiva de la tribu fore en Papúa-Nueva Guinea, que se manifestaba predominantemente por síntomas cerebelosos, con demencia ulterior, y que era sostenida por prácticas de antropofagia ritual (15). Si bien se presumía desaparecida, recientemente se describieron casos con una latencia de varias décadas hasta la aparición de los síntomas, que sugieren la posibilidad de la existencia de formas de muy larga latencia también para la vCJ(16).
En Uruguay existe una comisión de vigilancia de enfermedades priónicas en el animal y en el hombre creada en 1999 en la órbita del Ministerio de Salud Pública (MSP) y del Ministerio de Ganadería, Agricultura y Pesca, integrada también por el Instituto de Neurología de la Universidad de la República. Las enfermedades priónicas en el hombre son de denuncia obligatoria ante el MSP desde 1997. El decreto 64/004 ratificó dicha obligatoriedad(17).
En este trabajo se describe la realidad epidemiológica de estas enfermedades en Uruguay así como sus principales características clínicas y paraclínicas.
Material y método
Se realizó un estudio de cohorte retroprospectiva de casos clínicos de enfermedades priónicas diagnosticadas en nuestro país en el período 1984-2009 inclusive. La captación de casos se realizó mediante la recolección de información proveniente de neurólogos, neurofisiólogos, genetistas, certificados de defunción aportados por la División Epidemiología del MSP, estudios de LCR para la proteína 14-3-3 realizados en nuestro medio y el exterior.
Se utilizaron los criterios diagnósticos definidos por la OMS (anexo 1)(18).
Para la recolección de datos se utilizó una planilla especialmente diseñada, modificación de la planilla del Centro de Vigilancia Epidemiológica en ECJ de Argentina (19). Dicha planilla se completó junto con el neurólogo tratante, revisando la historia clínica y en muchas oportunidades mediante el examen clínico directo por parte de algún integrante del equipo investigador.
Referente al análisis estadístico se utilizó el programa SPSS (versión 17.0). Para el análisis descriptivo se utilizaron como medidas de resumen: a) para las variables continua media, desvío estándar, mediana; b) para las variables categóricas proporciones.
Para el análisis comparativo se utilizaron según: a) tipo y cantidad de variables a comparar, y b) número de efectivos y de grupos a comparar los siguientes tests: test de T (test paramétrico) y los tests de chi cuadrado o test de Fisher (según los efectivos teóricos, tests no paramétricos).
Para el análisis de sobrevida se utilizó el método de Kaplan-Meier y el test de Log Rank; dado el escaso número de formas familiares, se consideraron en conjunto ambas mutaciones.
Los tests estadísticos fueron realizados con un nivel de significación de 95% (riesgo alfa = 0,05).
Resultados
Se lograron identificar 42 casos de ECJ en Uruguay en el período mencionado.
La fuente de datos fue aportada fundamentalmente por parte de neurólogos, neurofisiólogos y la División Epidemiología del MSP.
En cuanto a las fuentes de procedencia de los casos clínicos, 55% correspondieron a instituciones de asistencia médica colectiva (IAMC, subsector privado); 40% a instituciones de asistencia pública (hospitales del MSP, Hospital de Clínicas, Hospital Militar y Hospital Policial), mientras que en 5% no se aportaron datos sobre la procedencia de los pacientes.
Se reviso la literatura nacional no encontrándose ningún caso descripto de enfermedad prionica hasta el fin del año 2009; en la literatura internacional se describió la existencia de una familia uruguaya con ECJ con una nueva mutación.
Se registraron 20 casos de sexo masculino y 22 casos de sexo femenino, que corresponden a 47,6% y a 52,4%, respectivamente, de la muestra.
Para la ECJ total, la distribución por período (en 1997 se inició la denuncia obligatoria de la enfermedad), considerando todas las categorías diagnósticas, para el período 1984-1996 se captaron 11 casos (promedio anual: 1) y para el período 1997-2009 se captaron 31 casos (promedio anual: 2,4).
Tomando únicamente las formas esporádicas en el primer período hubo ocho casos (promedio anual: 0,73) y en el segundo, 26 casos (promedio anual: 2).
La tasa de incidencia global estimada (considerando solo formas probables y definitivas) desde 1997 a 2009 en Uruguay fue de 0,7 casos por 1.000.000 de habitantes por año.
La tasa de incidencia para la forma esporádica (considerando solo formas probables y definitivas) desde 1997 a 2009 en Uruguay fue de 0,64 casos por 1.000.000 de habitantes por año.
Con respecto a las formas de ECJ se contabilizaron 34 (81%) casos de ECJ esporádico (81%) y 8 casos (19%) de ECJ hereditario (figura 1).
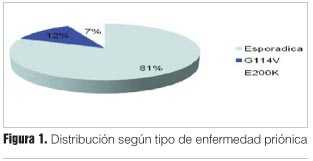
No se constataron en nuestro trabajo otras formas de enfermedades priónicas.
En cuanto a la distribución etaria, la ECJ forma hereditaria tuvo una media de 28,6 años (DS 13,1 años) en su aparición para la mutación G114V y de 65,7 años (DS 2,5 años) para la mutación E200K; mientras que para la ECJ forma esporádica tuvo una media de 61,6 años en su edad de aparición, con un DS de 2,5 años.
La diferencia en la edad de presentación entre las formas esporádicas y hereditaria fue significativa desde el punto de vista estadístico (p > 0,0001, test de ANOVA) (figura 2).
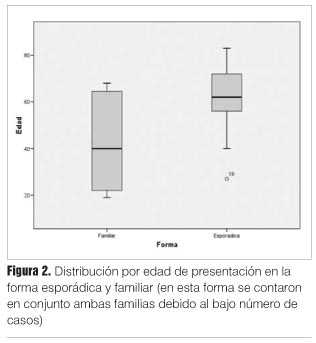
Para el sexo masculino el promedio de edad de presentación de ECJ esporádica fue de 61,7 años (DS 14,1), en tanto para el sexo femenino el promedio de presentación fue de 61,4 años (DS 9,9).
No se hallaron diferencias estadísticamente significativas entre las edades de ambos sexos.
Con respecto a los niveles de confirmación diagnóstica nos referiremos exclusivamente a los casos de ECJ esporádicos (dado que los casos correspondientes a las ECJ hereditarias están sellados sus diagnósticos por medio de estudios anátomo-patológicos y genéticos); de los 34 casos registrados, dos casos (5,9%) correspondieron a ECJ con criterio definitivo, 28 casos (82,4%) a formas probables y 4 casos (11,8%) a formas posibles (figura 3).
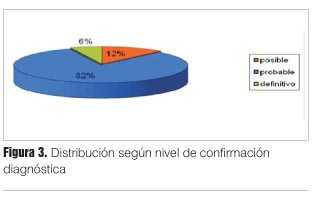
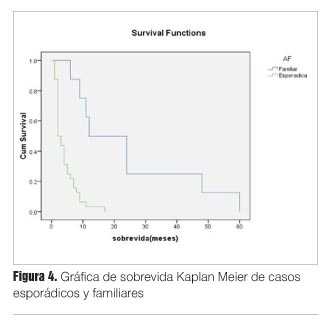
Referente al análisis de sobrevida las formas esporádicas presentaron una sobrevida significativamente menor con respecto a la formas familiares (mediana de sobrevida 2 meses versus 12 meses, respectivamente).
Para este análisis se consideraron en conjunto ambas formas familiares (mediana de sobrevida para G114V, 24 meses y para E200K, 11 meses) (figura 4).
Presentación clínica
Destacamos los síntomas-signos y síndromes más frecuentes encontrados en nuestra serie de pacientes, incluyendo en este análisis descriptivo solamente las formas esporádicas.
Los síntomas y signos más frecuentes fueron: demencia de rápida progresión (100%), signos extrapiramidales (94%), mioclonias (94%), signos piramidales (76%), signos cerebelosos (53%), mutismo aquinético (58%) y alteraciones visuales (15%).
Paraclínica
EEG
Al 90% de los pacientes se les realizó uno o más EEG durante la evolución siendo todos patológicos.
De estos, 45% presentó complejos periódicos (EEG típico), el resto presentó alteraciones atípicas (enlentecimiento difuso) (figuras 5 y 6).
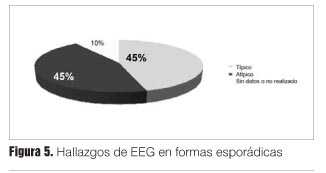
En cuanto a las formas familiares: los pacientes con la mutación G114V no mostraron complejos periódicos y de los pacientes con la mutación E200K se encontraron complejos periódicos en uno.
Proteína 14-3-3
En las formas esporádicas se realizó la búsqueda de la misma en 52,4% de los pacientes siendo positiva en 91% de los casos.
En las formas familiares se realizó solamente en un paciente de los tres con mutación E200K y fue negativa, fue realizada en un paciente con la mutación G114V y fue también negativa.
RM encefálica
Fue realizada a 20% de los pacientes y de estos, 57% presentaron hallazgos compatibles con ECJ (figura 7).
Discusión
Prácticamente todos los datos presentados en este trabajo fueron aportados por neurólogos, neurofisiólogos y neuropatólogos. Dadas las características de la enfermedad y la distribución de neurólogos en todo el país, es posible realizar un relevamiento y una vigilancia epidemiológica bastante estricta de estas enfermedades en nuestro medio, especialmente en relación con el diagnóstico de posible y probable de la ECJ esporádica. No existen hasta el momento posibilidades de realizar un diagnóstico definitivo dado que no hay aún en nuestro país laboratorios de patología que cumplan con las estrictas medidas de bioseguridad que se requieren; los casos diagnosticados como definitivos fueron consecuencia de la realización de biopsia cerebral ante planteos de diagnósticos diferenciales potencialmente tratables.
No existieron diferencias significativas en relación con el sexo de los afectados en los 42 pacientes detectados, siendo igual la prevalencia para el sexo masculino y femenino tal como señala la literatura internacional (20,21).
Se observó un mayor índice de diagnóstico para el período 1995-2009 en relación con la década anterior. Seguramente esta disparidad corresponda a un incremento en el diagnóstico más que a un aumento de la incidencia de la enfermedad. Coincide con la aparición de la vCJ detectada en el Reino Unido, que popularizó las enfermedades priónicas, logrando que la comunidad médica y neurológica prestase mayor atención y agudeza en el diagnóstico de estas enfermedades.
La incidencia anual de nuevos casos de enfermedad de Creutzfeldt-Jakob fue de 0,7 casos por año por millón de habitantes, la cual es comparable a la frecuencia hallada a nivel mundial (que varía entre 0,5 a 1,5 nuevos casos por año por millón de habitantes)(7,21,22).
Los casos detectados incluyen formas esporádicas y familiares; destacamos que no existieron casos iatrogénicos ni de la variante.
Encontramos en nuestra serie una alta proporción de casos familiares siendo estos de 19,4%, el 80,6% restante correspondió a casos esporádicos; esta distribución es diferente a la encontrada en otras series donde la frecuencia de casos familiares es menor: de 10% a 15%. De todas formas, dado el escaso número de nuestros pacientes, no podemos sacar conclusiones significativas.
La media de edad de presentación para los casos esporádicos fue de 61,6 años con un DS de 2,5, lo cual es similar a la media de edad de presentación para esta patología en la literatura consultada, que es de 61 años. La edad media de presentación de los casos familiares fue inferior para la mutación G114V (28,6 años) y similar a la esporádica en el caso de la mutación E200K (65,7 años, coincidente con la literatura)(21,22).
Destacamos que la mutación G114V fue en su momento una mutación inédita y reconocida por primera vez en una familia uruguaya, lo que ameritó la publicación en una revista internacional(23).
En cuanto a la seguridad del diagnóstico, determinada según criterios de la OMS, encontramos que en la mayoría de los pacientes (82%) el diagnóstico fue de probable, este se acerca en 90% al diagnóstico de certeza(24).
En nuestra serie tuvimos un bajo porcentaje de diagnósticos definitivos (6%), dado que la anatomía patológica se realizó solo en pacientes en los cuales no se sospechaba el diagnóstico de enfermedad priónica.
Sumando los casos probables y definitivos obtenemos que 88% de nuestros pacientes tuvieron una alta certeza diagnóstica(7,22,24).
La comparación de las curvas de sobrevida de los casos familiares (12 meses) y esporádicos (2 meses) fueron estadísticamente significativas (p = 0,0001), lo cual es esperable dado que la forma familiar se caracteriza por presentar una sobrevida mucho más larga que las formas esporádicas.
La mediana de sobrevida de los casos esporádicos fue más corta que la descripta en otras series que incluyen un número mayor de pacientes en la cual la mediana es de cinco meses(7, 20,21).
Las formas clínicas de presentación y evolución fueron similares a las descriptas en otras series(25-27).
Del análisis de los estudios paraclínicos realizados a nuestros pacientes encontramos que fueron de alto rendimiento diagnóstico los siguientes.
EEG: fue realizado en la mayoría de los pacientes (90%) pero de forma no monitoreada; dentro de estos encontramos que 45% presentaron un trazado típico, el resto presentó alteraciones inespecíficas. Destacamos que ningún paciente a los cuales se les realizó este estudio presentó un trazado normal.
Proteína 14-3-3: se realizó en 52,4% de los pacientes, dentro de estos encontramos que la determinación fue positiva en 91% de los casos.
RM de cráneo: se realizó en 20% de los pacientes, de estos estudios, 57% presentaron alteraciones típicas de ECJ esporádica. El bajo porcentaje de estudios realizados se debe a que esta técnica fue de difícil acceso en la secuencia de difusión hasta fines de la década de 1990.
Los hallazgos imagenológicos (RM encefálica) no están incluidos como criterio diagnóstico, pero los hallazgos de la misma son altamente sugerentes de ECJ (20,25,28).
Este trabajo se encuentra comprendido en un esfuerzo conjunto que realiza el Instituto de Neurología y la División Epidemiología del MSP en el monitoreo de las enfermedades priónicas en nuestro país desde varios años. Si bien se trata de enfermedades de baja frecuencia, la detección de posibles "clusters" o la eventualidad de determinadas atipías en su presentación llevan a focalizar la atención clínico-epidemiológica, para evitar consecuencias deletéreas en nuestra economía nacional basada en la ganadería.
Agradecimiento
Este trabajo fue posible gracias a la información, el aporte y el apoyo brindado por los siguientes colegas: Dres. J. L. Ardanaz, S. Bonnevaux, J. Caamaño, L. Carrasco, M. Castagnola, J. Favat +, D. Cibils, M. Fernández, S. Gianarelli, M. Gnocchi, C. Legnani, M. Leonardi, I. Lezama, C. Martínez Collet, C. Oheninger, L. Rosa (División Epidemiología del MSP), L. Ponce, A. L. Taratuto (Argentina), L. Vernengo, C. Volonté, R. Xavier, I. Zerrr (Alemania), C. López, N. Sánchez, M. Fariña, L. Ferix, J. Coehlo, Lic. Mónica Castro (División Epidemiología del MSP), Sra. M. Rauch (bibliotecóloga).
Summary
Introduction: prion diseases are progressive neurodegenerative disorders that may result in death and are distinguished by long incubation periods before presenting a clinical manifestation. Today they are scientifically interesting since they follow a protein auto-replicant model without the participation of nucleic acids.
These diseases appear sporadically, they are hereditary and infectious as well. The appearance of bovine spongiform encephalopathy and its consequences on human beings and Creutzfeldt-Jacob disease variations reinforce the need for a strict epidemiological control in the field.
Objective: to describe the reality of these diseases in Uruguay from 1984 through 2009.
Method: we conducted a retrospective, descriptive review of clinical cases of prion diseases diagnosed in our country.
Results: we managed to identify 43 cases of Creutzfeldt-Jacob disease in Uruguay (8 hereditary forms and 34 sporadic forms). Global incidence estimated rate was 0.7 cases per million inhabitants, considering both probable and definitive forms.
Conclusions: we have exclusively identified sporadic and hereditary cases of Creutzfeldt-Jacob disease, we have not identified variations in connection with bovine spongiform encephalopathy or other kinds of prion diseases affecting human beings.
Given the characteristics of the disease and the distribution of neurologists in the country enable a rather strict survey and epidemiological surveillance of these diseases in our country.
Resumo
Introdução: as doenças priônicas são enfermidades de caráter degenerativo do sistema nervoso central de curso progressivo y desenlace fatal que apresentam longos períodos de incubação antes de manifestar-se clinicamente sendo hoje de grande interesse científico, pois respondem a um modelo autorreplicante proteico sem intervenção de ácidos nucleicos. Têm ademais um caráter simultâneo de aparição esporádica, hereditária e infectante. O aparecimento da encefalopatia bovina espongiforme, a variante da doença de Creutzfeldt-Jacob e suas consequências nos seres humanos, destacam a necessidade de um controle epidemiológico estrito.
Objetivo: descrever a realidade destas doenças no Uruguai no período 1984-2009 inclusive.
Material e método: realizou-se uma revisão descritiva y retrospectiva de casos clínicos das doenças priônicas diagnosticadas no nosso pais.
Resultados: identificaram-se 42 casos de doença de Creutzfeldt-Jakob no Uruguai (8 formas hereditárias e 34 formas esporádicas). A taxa de incidência global estimada foi de 0,7 casos por milhão de habitantes por ano, considerando as formas prováveis e definitivas.
Conclusões: neste trabalho detectaram-se exclusivamente os casos esporádicos e familiares de Creutzfeldt-Jacob; não se identificaram casos vinculados à variante relacionada com a encefalopatia espongiforme bovina, nem a outros tipos de doenças priônicas que afetam a los seres humanos.
Considerando as características desta patologia e a distribuição de neurologistas no país, é possível realizar um relevamento e uma vigilância epidemiológica bastante estrita de estas doenças no nosso meio.
Bibliografía
1. Prusiner SB. Shattek lecture-neurodegenerative diseases and prions. N Engl J Med 2001; 334(20): 1516-26.
2. Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, et al. Conversion of alfa-helices into beta sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA 1993; 90(23): 10962-6.
3. McKinley MP, Meyer RK, Kenaga L, Rahbar F, Cotter R, Serban A, et al. Scrapie Prion Rod formation "in vitro" requieres both detergent and limited proteolisis. J Virol 1991; 65(3): 1340-51.
4. Salamano R, Cibils D. Aspectos clínicos y paraclínicos de las enfermedades priónicas. In: Salamano R. Neurovirosis y enfermedades priónicas. Montevideo: Oficina del Libro Fefmur, 2002: 155-62.
5. Rodríguez Escanlar M. Genética y enfermedades priónicas. In: Salamano R. Neurovirosis y enfermedades priónicas. Montevideo: Oficina del Libro Fefmur, 2002: 163-75.
6. Perdomo E. Introducción enfermedades priónicas. In: Salamano R. Neurovirosis y enfermedades priónicas. Montevideo: Oficina del Libro Fefmur, 2002: 137-42.
7. Zivkovic S, Boada M, López O. Revisión de la enfermmedad de Creutzfeldt-Jakob y otras enfermedades priónicas. Rev Neurol 2000; 31(12): 1171-9.
8. Pauri F, Amabile G, Fattapposta F, Pierallini A, Bianco F. Sporadic Creutzfeldt-Jakob disease without dementia at onset: clinical features, laboratory tests and sequential diffusion MRI (in an autopsy-proven case). Neurol Sci 2004; 25(4): 234-7.
9. Meissner B, Körtner K, Bartl M, Jastrow U, Mollenhauer B, Schröter A, et al. Sporadic Creutzfeldt-Jakob disease magnetic resonance imaging and clinical findings. Neurology 2004; 63(3): 450-6.
10. Wolfe K, Leach JL, Kissela B, Fortuna RB. A patient with rapidly progressive mental status decline: imaging of Creutzfeldt-Jakob disease. Infect Dis Clin Pract 2006; 14(3): 161-5.
11. Prusiner SB, HSiao KK, Bredesen DE, De Armond SJ. Prion disease. In: Vinken PJ, Bruyn GW, Klawans HL, McKendall RR, eds. Viral disease: handbook of clinical Neurology, Vol 12(56). Amsterdam: Elsevier, 1989: 543-80.
12. The University of Edinburgh. The National Creutzfeldt-Jakob Disease Research & Surveillance Unit (NCJDRSU). Information on variant CJD. Disponible en: http://www.cjd.ed.ac.uk/ [Consulta: 4/6/2011].
13. Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A, et al. new variant of Creutzfeldt-Jacob disease in the UK. Lancet 1996; 347(9006): 921-5.
14. Collinge J. Variant Creutzfeldt-Jacob disease. Lancet 1999; 354(9175): 317-23.
15. Gajdusek DC, Zigas V. Degenerative disease of the central nervous system in New-Guinea: the endemic ocurrence of kuru in the native population. N Engl J Med 1957; 257(20): 974-8.
16. Collinge J, Whitfield J, McKintosh E, Beck J, Mead S, Thomas DJ, et al. Kuru in the 21st century: an acquired human prion disease with very long incubation periods. Lancet 2006; 367(9528): 2068-74.
17. Decreto Nº 64/004 - Código Nacional sobre Enfermedades y Eventos Sanitarios de Notificación Obligatoria. Montevideo, 18 de febrero de 2004. Disponible en: http://www.elderechodigital.com.uy/smu/legisla/D0400064.html [Consulta: 19/5/ 2011].
18. World Health Organization. Global surveillance, diagnosis and therapy of human transmissible spongiform encephalopathies: Report of a WHO Consultation. Geneva: WHO, 1998. Disponible en: http://whqlibdoc.who.int/hq/1998/ WHO_EMC_ZDI_98.9.pdf [Consulta:19/5/2011].
19. Formulario de notificación de enfermedad de Creutzfeld-Jakob y otras relacionadas. Boletín Digital 2006, No 1. Disponible en: http://www.sociedadpanamericananeurologia.org [Consulta: 19/5/2011].
20. Blumenkron D, Guerrero P, Ramiro M. Enfermedad de Creutzfeldt-Jakob. Med Int Mex 2007; 23(1): 34-46.
21. Collins S, Boyd A, Lee JS, Lewis V, Fletcher A, McLean CA, et al. Creutzfeldt-Jakob disease in Australia 1970-1999. Neurology 2002; 59(9): 1365-71.
22. Begué C, Piccardo P, Taratuto AL. Encefalopatías espongiformes transmisibles: enfermedad de Creutzfeldt-Jakob. In: Salamano R, Scavone C, Savio E, Wajskopf S. Neuroinfecciones en el adulto y el niño. Montevideo: Arena, 2008: 203-13.
23. Rodriguez MM, Peoc'h K, Haïk S, Bouchet C, Vernengo L, Mañana G, et al. A novel mutation (G114V) in the prion protein gene in a family with inherited prion disease. Neurology 2005; 64(8): 1455-7.
24. Zerr I, Pocchiari M, Collins S, Brandel JP, de Pedro Cuesta J, Knight RS, et al. Analysis of EEG and CSF 14-3-3 proteins as aids to diagnosis of CJD. Neurology 2000; 55(6): 811-5.
25. Romero MJ, Romero J. La enfermedad de Creutzfeldt-Jakob esporádica: variaciones fenotípicas. Neurología 2002; 17(7):366-77.
26. Zerr I. Manifestaciones clínicas y test diagnósticos de las enfermedades priónicas en humanos. Arch Inst Neurol 2001; 4(1): 41-8.
27. Snowden JS, Mann DM, Neary D. Distinct neuropsychological characteristics in Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry 2002; 73(6): 686-94.
28. Geschwind M. Human prion Disease. American Academy of Neurology. ANN 59th Anual Meeting, May, 2007: 2FC.001-70-6.


{kind=link}
{kind=link}