










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkRevista Médica del Uruguay
versión On-line ISSN 1688-0390
Rev. Méd. Urug. vol.27 no.3 Montevideo set. 2011
Hipertensión pulmonar tromboembólica crónica
Juan Pablo Salisbury*, Pablo Curbelo†, Mercedes Arcaus*, Jorge Cáneva‡
Resumen
La hipertensión pulmonar tromboembólica crónica (HPTEC) es una entidad poco frecuente que se puede observar hasta en 4% de los sobrevivientes de un tromboembolismo pulmonar agudo (TEPA). Se trata de una enfermedad con un pobre pronóstico de no realizarse un diagnóstico precoz y un tratamiento oportuno. Paradójicamente se trata de la única forma de hipertensión pulmonar que cuenta con una posibilidad curativa mediante la endarterectomía pulmonar (EP). Su diagnóstico se establece inicialmente mediante métodos no invasivos como el ecocardiograma Doppler y el centellograma pulmonar ventilación perfusión (V/Q). La evaluación diagnóstica y preoperatoria se completa con un cateterismo cardíaco derecho y una angiografía pulmonar. Estos confirman el diagnóstico de HPTEC y determinan la posibilidad de la EP.
Palabras clave: HIPERTENSIÓN PULMONAR. TROMBOEMBOLIA. ENFERMEDAD CRÓNICA .
Keywords: HYPERTENSION, PULMONARY. THROMBOEMBOLISM. CHRONIC DISEASE.
* Asistentes de la Cátedra de Neumología. Hospital Maciel. Facultad de Medicina. Universidad de la República. Uruguay.
† Profesor Adjunto Cátedra de Neumología. Hospital Maciel. Facultad de Medicina. Universidad de la República. Uruguay.
‡ Profesor Titular del Departamento de Clínica Médica. Universidad Favaloro. Fundación Favaloro. Buenos Aires. Argentina
Correspondencia: Dr. Juan Pablo Salisbury
Francisco Araucho 1166 Ap. 5. Montevideo, Uruguay
Correo electrónico: jpsalis@hotmail.com
Recibido: 7/2/11.
Aceptado: 27/6/11.
Introducción
La hipertensión pulmonar tromboembólica crónica (HPTEC) es una enfermedad poco conocida que se produce por la oclusión crónica de las arterias pulmonares por trombos organizados(1). Su consecuencia es el aumento de la resistencia vascular pulmonar (RVP) determinando la hipertensión pulmonar (HP) y que evoluciona a la claudicación del ventrículo derecho y a la muerte de no tener un diagnóstico precoz y un tratamiento adecuado(1-3). Su frecuencia es difícil de estimar debido a que se trata de una entidad subdiagnosticada debido a su baja sospecha y difícil diagnóstico, ya que sus síntomas son inespecíficos y en más de la mitad de los casos no se detectan episodios previos de tromboembolismo pulmonar agudo (TEPA)(1).
La herramienta inicial para diagnosticar la etiología tromboembólica de la HP es el centellograma pulmonar ventilación perfusión(4). Un resultado positivo obliga a completar con una angiotomografía multicorte o una angiografía pulmonar convencional. Esta última confirma el diagnóstico y determina la viabilidad de la endarterectomía pulmonar (EP), único tratamiento curativo para esta patología(5-7). Dicho procedimiento debe realizarse en un centro de referencia con experiencia en la técnica.
El cateterismo cardíaco derecho (CCD) confirma el diagnóstico, severidad y otorga elementos pronósticos de la HPTEC(5).
Epidemiología
Su real incidencia es muy difícil de estimar, ya que su etiopatogenia e historia natural son poco conocidas. Si bien el desarrollo de HPTEC guarda una relación etiopatogénica con la presencia previa de TEPA, más de 50% de los pacientes diagnosticados no presentan antecedentes de TEPA clínicamente documentados, por lo que es probable que la incidencia de esta enfermedad sea mayor que la estimable a partir de la de TEPA(1).
Originalmente se creía que 0,1% a 0,5% de los pacientes que sobreviven a un episodio de TEPA desarrollarían HPTEC(8).
Pengo y colaboradores publicaron en el año 2004 un estudio prospectivo en el que se realizó un seguimiento a 223 pacientes que sobrevivieron a un TEPA, durante un período promedio de 94 meses. Encontraron una incidencia acumulada de HPTEC de 1% a los seis meses, 3,1% al año y 3,8% a los dos años. Siendo los principales factores de riesgo asociados identificados: el antecedente de TEPA previo, un defecto mayor en la perfusión (segmentario) del centellograma, la etiología idiopática del mismo y la edad joven. Concluyeron que la HPTEC es una complicación relativamente común y grave del TEPA, lo cual obliga a realizar un seguimiento sistemático de estos pacientes luego del evento agudo(9).
Definición y clasificación
La HP es un estado hemodinámico y fisiopatológico común a muchas enfermedades clínicas, las cuales han sido clasificadas en cinco grupos en el último consenso realizado en Dana Point, California, en el año 2008 (tabla 1)(5). La HPTEC integra el grupo 4 de la nueva clasificación. La hipertensión arterial pulmonar (HAP) precapilar se define como un aumento de la presión arterial pulmonar media = 25 mmHg, presión capilar pulmonar = 15 mmHg y resistencia vascular pulmonar > 2 unidades Wood evaluada mediante CCD(5).
El diagnóstico de HPTEC se establece por la presencia de HAP en pacientes con trombos oclusivos, crónicos y organizados en las arterias pulmonares persistentes luego de tres meses del episodio agudo de TEPA. Asimismo, se puede arribar al diagnóstico de HPTEC en un paciente sin antecedentes de TEPA conocido durante la evaluación de un caso de HP de etiología no aclarada. Se trata de la única forma de HP potencialmente curable mediante cirugía; de lo contrario evoluciona a la insuficiencia cardíaca derecha y a la muerte.
Etiopatogenia
La patogenia de la HPTEC no se conoce por completo. Si bien se considera un trastorno secundario a un tromboembolismo venoso (TEV), frecuentemente no se encuentra asociación con trombosis venosa profunda ni con los factores de riesgo protrombóticos clásicos(5). La historia natural del TEPA es la evolución a la resolución espontánea, normalizando las alteraciones hemodinámicas y los fenómenos trombóticos en un plazo aproximado de 30 días mediante la trombolisis endógena. Las razones para la resolución incompleta de la embolia pulmonar no han sido identificadas. Si bien el lecho vascular pulmonar normal presenta un elevado potencial fibrinolítico, su alteración y contribución al desarrollo de HPTEC no han podido ser corroboradas.
Diferentes estudios han intentado vincular, sin éxito, a los factores protrombóticos con la HPTEC; sin embargo, no se ha encontrado asociación significativa con factores de riesgo protrombóticos hereditarios entre los pacientes con HAP y los sujetos control(3). En contraste, los únicos elementos que se han relacionado con HPTEC hasta el momento son los anticuerpos anticardiolipina, que se encuentran en 10% a 20% de los pacientes y niveles elevados de factor VIII(10,11).
A pesar de la evidencia que une al TEPA con la HPTEC, existen otras hipótesis alternativas sobre su patogenia que sugieren una arteriopatía primaria o secundaria a trombosis in situ como causas de la oclusión vascular pulmonar(3).
En estos casos, la enfermedad probablemente comience por lesiones trombóticas o inflamatorias en la propia vasculatura pulmonar (trombosis in situ). Una vez que se produce la lesión del vaso y aumenta la presión endovascular, se iniciaría un proceso de remodelado vascular que se autoperpetuaría evolucionando hacia la HPTEC. Varios autores proponen que este remodelado vascular puede ser perpetuado por el efecto de infecciones, fenómenos autoinmunes, enfermedades malignas o el tratamiento sustitutivo tiroideo(11-13).
Diferentes enfermedades se asocian a una mayor incidencia de HPTEC y peor pronóstico a saber: la esplenectomía, la derivación ventrículo atrial, marcapasos infectados, desórdenes mieloproliferativos, enfermedad inflamatoria intestinal y la osteomielitis crónica(10). Recientemente surgieron nuevos factores de riesgo para el desarrollo de esta forma de HP, como el tratamiento sustitutivo con levotiroxina y el antecedente de enfermedades neoplásicas(11).
Presentación clínica y diagnóstico
Los síntomas de HPTEC son intermitentes y se producen cuando resulta afectada más de 50% de la circulación pulmonar. La intolerancia al ejercicio y la disnea al esfuerzo son los síntomas más frecuentes junto al dolor torácico. El síncope y la hemoptisis constituyen síntomas que indican gravedad y mal pronóstico. A medida que la enfermedad progresa comienzan a objetivarse elementos clínicos de insuficiencia cardíaca derecha(1,5).
La evolución de la HPTEC es episódica, con períodos prolongados denominados de "luna de miel", en los que los síntomas son leves o inexistentes.
Muchos pacientes con HPTEC tienen el antecedente bien documentado de TEPA o TVP (trombosis venosa profunda), o ambos, mientras que en otros el evento pasa clínicamente desapercibido. Asimismo, la observación de que la gran mayoría de las personas que sufren un TEPA no desarrollan HPTEC sugiere que hay otros factores que son importantes en el desarrollo de la enfermedad(14). Por lo tanto, las principales guías de manejo de la HP sugieren que durante la evaluación de un paciente con HP, que presente el antecedente de TEPA o HP de etiología no aclarada, debe estudiarse la etiología tromboembólica(5,14).
Al examen físico se puede encontrar un latido palpable amplio y sostenido a nivel del segundo espacio intercostal izquierdo, traducción de la dilatación de la arteria pulmonar debido a la sobrecarga de presión en la misma, acompañada a la auscultación de un segundo tono aumentado en foco pulmonar, resultante del cierre violento de la válvula pulmonar como respuesta a una presión diastólica elevada a dicho nivel. Todos estos elementos conforman el complejo de la pulmonar de Chávez.
A la sospecha de HP que surge de la historia clínica y el examen físico se le deben sumar los aportes de la radiografía de tórax (RxTx) y del electrocardiograma (ECG). La RxTx se encuentra alterada en 90% de los casos al momento del diagnóstico, siendo las principales alteraciones el aumento de calibre del tronco de la arteria pulmonar (rectificación o abombamiento del tronco de la arteria pulmonar), aumento de calibre de las arterias lobares y la oligohemia periférica, junto con la remodelación de la silueta cardíaca a predominio derecho. El ECG presenta una baja sensibilidad diagnóstica, siendo los elementos más frecuentes la dilatación y/o hipertrofia del ventrículo derecho (87%), y la desviación del eje a derecha (79%)(5).
El ecocardiograma es la técnica de elección para screening y detección precoz de la enfermedad.
El principal factor de riesgo, como ha sido mencionado, es el TEPA, por lo tanto se recomienda que estos pacientes sean evaluados con un ecocardiograma a los tres y seis meses y luego anualmente durante al menos los dos primeros años en busca de esta complicación(10). La presencia de una presión arterial pulmonar sistólica (PAPs) superior a 50 mmHg al momento del diagnóstico de TEPA, es altamente probable su persistencia al año del mismo(2).
Ante la sospecha etiológica tromboembólica se recomienda inicialmente realizar un centellograma pulmonar ventilación perfusión, ya que es el método de diagnóstico más recomendado debido a su alta sensibilidad(4). Un centellograma pulmonar normal o de baja probabilidad excluye eficazmente la HPTEC con una sensibilidad de 90% a 100% y una especificidad de 94% a 100%(4,5).
La presencia de un defecto segmentario o múltiples defectos segmentarios o subsegmentarios en la perfusión hace el diagnóstico de HPTEC más probable, aunque existen otras condiciones como la enfermedad veno-oclusiva pulmonar (EVOP) o la hemangiomatosis capilar pulmonar (HCP), las que deben ser tenidas en cuenta como diagnósticos diferenciales.
Si el resultado del centellograma pulmonar es indeterminado o revela defectos de perfusión, debe realizarse una angiotomografía helicoidal. Su importancia radica en poder observar la topografía y extensión de los trombos a nivel del árbol arterial pulmonar, ya que la topografía central de los mismos los hace pasibles de ser resecados quirúrgicamente mediante la EP.
En la actualidad los nuevos tomógrafos multicortes y la reconstrucción en tres dimensiones nos permiten realizar una evaluación con elevada calidad de imagen.
Sin embargo, la arteriografía convencional es el método patrón que define mejor la anatomía lesional y permite la eventual planificación quirúrgica.
El CCD es necesario no solo para confirmar el diagnóstico y descartar diagnósticos diferenciales como la EVOP y la HP poscapilar, sino que también valora la gravedad del deterioro hemodinámico y marca el pronóstico(5, 11,12).
El diagnóstico final de la HPTEC se basa en la presencia de HP precapilar (PAP media = 25 mmHg, presión capilar de enclavamiento pulmonar (PCEP) = 15 mmHg, resistencia vascular pulmonar (RVP) > 3 unidades Wood) en presencia de múltiples trombos, oclusivos, crónicos y organizados en las arterias pulmonares elásticas (principal, lobares, segmentarias, subsegmentarias) (5).
Tratamiento
La HPTEC es una enfermedad grave, con una sobrevida a cinco años de 30% cuando la presión arterial pulmonar media (PAPm) es > 40 mmHg y de 10% cuando es superior 50 mmHg(11). Un estudio de 49 pacientes con HPTEC confirmada mediante CCD, tratados solo con anticoagulación, reportó una mortalidad de 90% a los tres años, dejando en evidencia que dicho tratamiento no ofrece posibilidad curativa(12).
Estos estudios demuestran que el tratamiento médico solo no es suficiente para mejorar la sobrevida. Los malos resultados con tratamiento médico junto con el carácter obstructivo de las lesiones llevaron hace 40 años a plantear el tratamiento quirúrgico de la misma. En julio de 1970 se llevó a cabo, en el hospital de San Diego, California, la primera EP con éxito(15). Desde entonces es el tratamiento de elección ya que es el único que ofrece una posibilidad curativa para esta enfermedad(7,16-18).
Esto se apoya en su baja mortalidad (4%-10%) sumado a los buenos resultados obtenidos, normalizando la capacidad de ejercicio y los parámetros hemodinámicos.
A largo plazo, alrededor de 75% de los pacientes sometidos a una EP presenta buenos resultados funcionales y cerca de la mitad de los pacientes recupera una correcta tolerancia al ejercicio(19).
Las guías de manejo de esta patología recomiendan no considerar a un paciente como inoperable mientras el caso no haya sido evaluado por un centro de referencia en el manejo médico quirúrgico de la HP. Se considera que un centro tiene experiencia si realiza al menos 20 EP por año con una mortalidad menor a 10%(5).
La oportunidad para dicho procedimiento no está del todo establecida, recomendándose un plazo mínimo de tres meses de anticoagulación desde el momento del diagnóstico.
Su fundamento es que la cirugía puede ser mucho más difícil y menos satisfactoria si el trombo no se ha organizado completamente.
Los pacientes deben recibir anticoagulación de por vida ajustados a una razón normalizada internacional (INR) entre 2 y 3(5). La justificación de la anticoagulación es la prevención de la recurrencia de episodios embólicos, ya que una vez que la HPTEC está plenamente establecida no debemos esperar una regresión significativa con dicho tratamiento. Esto también ha llevado a algunos centros a implantar un filtro de vena cava inferior en forma sistemática cuando se realiza el CCD(7,18).
Una alternativa quirúrgica para pacientes no pasibles de EP y refractarios al tratamiento médico es el trasplante pulmonar, aunque ha de considerarse siempre una terapéutica de segunda línea, pues la EP presenta menor mortalidad hospitalaria y mayor supervivencia a largo plazo, sin los inconvenientes derivados de la inmunodepresión y el rechazo del injerto(6).
El procedimiento consiste en la realización de una EP bilateral mediante esternotomía media con soporte de circulación extracorpórea y períodos de paro circulatorio intermitente en hipotermia profunda. Comienza con la apertura de la arteria pulmonar en su sector intrapericárdico y desde allí se sigue circularmente hasta las ramas arteriales lobares, segmentarias y subsegmentarias de cada lóbulo (figura 1).
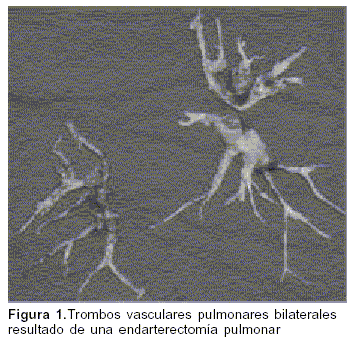
Debido a lo complejo del procedimiento se requiere de una profunda y exhaustiva evaluación en busca de realizar una selección idónea que garantice el éxito del procedimiento. En este sentido las variables más importantes son la topografía y extensión de los trombos, el nivel de RVP y las comorbilidades del paciente(7,20). Las indicaciones de EP se resumen en la tabla 2.

La única contraindicación absoluta de EP es la presencia de una severa enfermedad pulmonar subyacente. No hay un límite superior de RVP, grado de fallo ventricular derecho o de regurgitación tricuspídea que excluyan a un paciente de la opción quirúrgica(19).
Jamieson y colaboradores han demostrado que la localización de las lesiones halladas durante la cirugía tiene diferente pronóstico(7). Siguiendo la clasificación intraoperatoria propuesta por el mismo autor en el año 2003, se observó que las lesiones tipo I (presencia de trombos en arterias principales o lobares) se asocian con una mortalidad quirúrgica de 2,1%; las tipo II (engrosamiento de la íntima, fibrosis y bandas dentro de las arterias lobares sin trombos visibles) asocian una mortalidad de 5,3%; las tipo III (engrosamiento intimal, bandas fibrosas en ramas segmentarias y subsegmentarias sin trombos visibles) asocian una mortalidad de 5%, y las tipo IV (vasculopatía microscópica arteriolar distal) asocian una mortalidad quirúrgica de 25%(7). Sin embargo, como ya hemos mencionado, también existe una relación casi lineal entre la RVP preoperatoria y la mortalidad perioperatoria. En una serie francesa, la tasa de mortalidad reportada fue de 4% cuando la RVP preoperatoria fue < 900 dinas s.cm-5, pero aumentó a 10% en pacientes con RVP entre 900 y 1.200 dinas s.cm -5 y llegó a 20% en pacientes con RVP mayores(21). La HP residual posoperatoria se ha identificado como el predictor más importante de muerte. En una de las mayores series publicadas hasta la fecha por un centro de referencia en EP se reportó que los pacientes con RVP posoperatoria > 500 dinas s.cm- 5 tenían una mortalidad de 30,6% (15 de 49 pacientes), mientras que aquellos con RVP posoperatoria < 500 dinas s.cm- 5 tenían una mortalidad de 0,9% (4 de 434 pacientes) (7). Estos datos sugieren que no todos los pacientes con trombos centrales técnicamente resecables se benefician del tratamiento quirúrgico. Dartevelle y colaboradores han sugerido que los pacientes deben ser exhaustivamente seleccionados para EP y solo realizarla en aquellos pacientes en que se pueda predecir una reducción mayor a 50% de la RVP durante el preoperatorio(21). Esto obliga a realizar una exhaustiva valoración multidisciplinaria en busca de definir el candidato ideal para la misma. Por ejemplo, los pacientes con RVP desproporcionadamente altas, no explicadas por las lesiones oclusivas presentes en los estudios imagenológicos, es muy probable que asocien un alto grado de vasculopatía periférica (grupo 4 de la clasificación de Jamieson) y, por ende, un elevado riesgo perioperatorio y no se beneficien. Por lo tanto, la EP es el tratamiento de elección, aunque no siempre está indicada o disponible en el medio, como es el caso de nuestro país.
El tratamiento médico se encuentra indicado en las siguientes situaciones: 1) pacientes no aptos para la cirugía; 2) como puente a la EP con la finalidad de mejorar la hemodinamia pulmonar; 3) como tratamiento de la HP residual luego de la cirugía; 4) ante la negativa del paciente a realizarse un tratamiento quirúrgico. En los últimos años se han realizado diferentes ensayos clínicos con vasodilatadores pulmonares en la HPTEC no quirúrgica con algunos resultados alentadores.
El bosentán es un antagonista competitivo no selectivo de los receptores de endotelina-1 (ET-1). Presenta un efecto antiproliferativo a través de la inhibición de la producción de ET-1, la cual se concentra fundamentalmente a nivel arterial pulmonar.
El estudio BENEFIT (Bosentan Effects in Inoperable Forms of Chronic Thromboembolic Pulmonary Hypertension) fue el primer estudio aleatorizado, doble ciego, controlado por placebo en pacientes portadores de HPTEC inoperables o con HP persistente posquirúrgica. Demostró mejoría en un plazo de 16 semanas a nivel de la RVP (-24,1%, -193 dinas.s.cm-5; p=0,0001), índice cardíaco (IC) (+0,3 l/min/m2; p=0,0007) y reducción de los niveles de Pro-BNP (-622 ng/l; p=0,0034). Sin embargo, no demostró cambios en la clase funcional, distancia recorrida a la prueba de marcha de los 6 minutos (PM6M) ni en el tiempo a la aparición del deterioro clínico(22).
El sildenafil es un potente inhibidor selectivo de la fosfodiesterasa tipo 5 activo por vía oral que actúa a través de la vía del óxido nítrico incrementando los niveles de GMP cíclico ejerciendo un efecto vasodilatador y pudiendo también ejercer un efecto antiproliferativo a nivel del músculo liso vascular. Se trata de un fármaco ampliamente utilizado en pacientes con HPTEC debido a su bajo costo, fácil administración y mínimos efectos secundarios. Varios estudios han demostrado su eficacia en hipertensión arterial idiopática, asociada a enfermedad del tejido conectivo, cardiopatía congénita e hipertensión arterial tromboembólica crónica(23-26).
Un estudio con 19 pacientes portadores de HPTEC inoperables, randomizado, doble ciego, controlado con placebo que utilizó sildenafil por un período de 12 semanas no evidenció diferencia significativa en el objetivo primario (PM6M) entre ambos grupos, pero sí alcanzó varios de los objetivos secundarios mostrando mejorías significativas en el grupo de sildenafil en la clase funcional de la Organización Mundial de la Salud (OMS) y en la RVP(26). Sin embargo, en el seguimiento a 12 meses se observó mejoría significativa en la distancia recorrida, grado de disnea, niveles de pro-BNP y parámetros hemodinámicos (RVP, IC, PAPmedia, presión aurícula derecha). Esos datos sugieren que el sildenafil podría modificar favorablemente el pronóstico de estos pacientes.
Las prostaciclinas son un prostanoide natural, derivado del ácido araquidónico, producido por el endotelio vascular, tienen efecto vasodilatador, antiagregante, antiproliferativo y citoprotector. Los diferentes análogos sintéticos utilizados pueden ser administrados por vía oral, subcutánea, intravenosa o inhalada. Una infusión continua de prostaciclina fue la primera terapia demostrada que reduce la mortalidad en un estudio controlado en pacientes con HAP grave(27). Debido a los beneficios demostrados se comenzó a utilizar en pacientes con HPTEC. La prostaciclina primariamente utilizada en HPTEC es el epoprostenol intravenoso en perfusión continua. Se utilizó en pacientes en clase funcional (CF) de la New York Heart Association (NYHA) III-IV, sin indicación de cirugía, en la HP persistente posEP o como terapia puente a la misma o al trasplante pulmonar. Los resultados obtenidos luego de tratar a 23 pacientes con HPTEC inoperables y cuatro pacientes con HP poscirugía por tres meses fueron la mejoría en la clase funcional, distancia recorrida (TM6M + 66 metros) y perfil hemodinámico (reducción de 21% RVP). La supervivencia obtenida fue de 73%, 59% y 41% en el primer, segundo y tercer trimestre(28).
El iloprost es un análogo estable de la prostaciclina, que puede emplearse por vía intravenosa o inhalada. Su acción vasodilatadora es más duradera que la del epoprostenol. Su acción no se limita solo a la relajación de la vasculatura lisa pulmonar, también es inhibidor de la agregación plaquetaria, mejora el aclaramiento pulmonar de la endotelina 1, revierte el remodelado vascular e inhibe la proliferación y migración celular vascular(29).
Un ensayo multicéntrico, aleatorizado y controlado valoró la eficacia del iloprost inhalado en diversas formas de HP. El mismo incluyó un total de 203 pacientes randomizados en dos grupos, con un seguimiento a 12 semanas. En el grupo randomizados a recibir iloprost se incluyeron 101 pacientes, de los cuales 51 eran HP idiopática y los restantes 50 portadores de HP no idiopática, dentro de los cuales 33 eran de naturaleza tromboembólica(29).
Los resultados de este ensayo demostraron que el iloprost por vía inhalatoria logró una mejora clínicamente significativa en el objetivo primario (PM6M), así como beneficio en los objetivos secundarios (capacidad de ejercicio, CF de la NYHA y el deterioro clínico). Al igual que otros investigadores, los autores encontraron que el beneficio era mayor entre los pacientes con HAP idiopática y fue similar a los obtenidos con epoprostenol(27,28) y bosentán(30). Los autores concluyeron que iloprost inhalado es un fármaco seguro y eficaz para el tratamiento de la HAP grave y determinadas formas de HP dentro de las que se encuentra la HPTEC.
Pronóstico
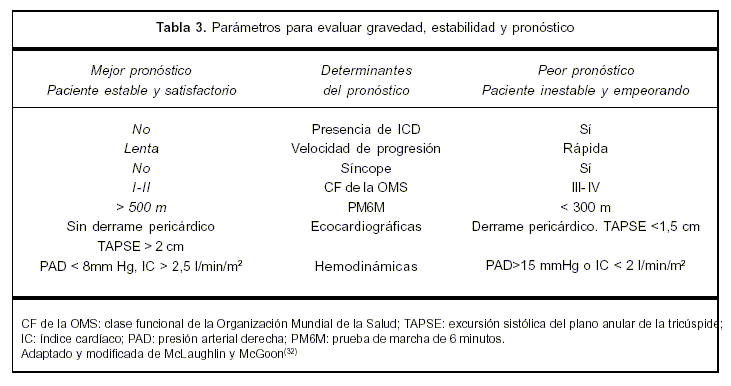
La evaluación y el seguimiento de los pacientes debe centrarse en variables con importancia pronóstica confirmada (tabla 3). La decisión acerca del tratamiento se debe de tomar en base a parámetros que reflejen los síntomas y la capacidad de ejercicio. En este sentido uno de los más simples, pero tal vez el más poderoso indicador de supervivencia es la clase funcional de la OMS. Las tres variables independientes de muerte asociadas a HPTEC no quirúrgicas son la presencia de enfermedad pulmonar obstructiva crónica, la mala tolerancia al ejercicio y la presencia de una PAP media mayor a 50 mmHg(31).
Situación en Uruguay
Nuestro país no cuenta con ningún centro con experiencia en EP, por lo cual se debería recurrir a un centro regional de referencia para el tratamiento definitivo de esta grave afección. Recientemente se ha logrado la financiación del CCD y se ha universalizado la cobertura con fármacos específicos en HP (iloprost inhalatorio, bosentan oral) que pueden ser usados en pacientes sin criterio para EP. Por otro lado, Uruguay cuenta con un programa de trasplante pulmonar en convenio con un centro regional argentino (Fundación Favaloro), indicado solo en casos no pasibles de EP y refractarios al tratamiento médico.
Conclusiones
La HPTEC es una de las etiologías de HP e integra el grupo de 4 de la clasificación actual de Dana Point patrocinada por la OMS. Se debe de tener una elevada sospecha clínica, debiéndose pensar en dicha etiología en el estudio de un paciente con HP de origen no aclarado. Se trata de la única causa de HP curable mediante cirugía, por lo que el pronóstico dependerá de contar con los requisitos necesarios para la misma.
El tratamiento médico actualmente logra mejorar la sintomatología del paciente y la hemodinamia en forma transitoria evolucionando a la insuficiencia cardíaca derecha y a la muerte cuanto mayor es la PAP media. Se necesitan trabajos prospectivos y randomizados con diferentes subgrupos de HPTEC y los diferentes fármacos existentes en la actualidad para poder contar con mayores datos acerca del tratamiento médico en esta enfermedad.
Summary
Chronic thromboembolic pulmonary hypertension (CTEPH) is an unusual entity that may be observed in up to 4% of patients surviving an acute pulmonary thromboembolism (APTE). It is a disease with a poor prognosis unless there is an early diagnosis and a timely treatment. Paradoxically, it is the only form of pulmonary hypertension that may be cured by means of a pulmonary endarterectomy (PE). It is initially diagnosed by non-invasive methods such as the Doppler echocardiogram and the ventilation/perfusion pulmonary scintigraphy (V/Q). Diagnostic and preoperative assessment is completed with a right cardiac catheterization and a pulmonary angiography. These techniques confirm the chronic thromboembolic pulmonary hypertension and define whether it is possible to perform a pulmonary endarterectomy.
Resumo
O tromboembolismo pulmonar crônico hipertensivo (TEPCH) é uma entidade pouco freqüente que se pode observar em até 4% dos sobreviventes de um tromboembolismo pulmonar agudo (TEPA). É uma patologia com mal prognóstico se não se realiza um diagnóstico precoce e tratamento oportuno. Paradoxalmente é a única forma de hipertensão pulmonar com possibilidade de cura pela endarterectomía pulmonar (EP). Seu diagnóstico é feito inicialmente empregando métodos não invasivos como o ecocardiografia bidimensional Doppler e a cintilografia de ventilação/perfusão pulmonar. Uma avaliação diagnóstica e pré-operatória completa inclui cateterismo cardíaco direito e angiografia pulmonar. Estes exames confirmam o diagnóstico de HPTEC e determinam a possibilidade de realizar uma EP.
Bibliografía
1. Lang IM. Chronic thromboembolic pulmonary hypertension- not so rare after all. N Engl J Med 2004; 350(22): 2236-8.
2. Tapson VF, Humbert M. Incidence and prevalence of chronic thromboembolic pulmonary hypertension: from acute to chronic pulmonary embolism. Proc Am Thorac Soc 2006; 3(7): 564-7.
3. Wolf M, Boyer-Neumann C, Parent F, Eschwege V, Jaillet H, Meyer D, et al. Thrombotic risk factors in pulmonary hypertension. Eur Respir J 2000; 15(2): 395-9.
4. Tunariu N, Gibbs SJ, Win Z, Gin-Sing W, Graham A, Gishen P, et al. Ventilation-perfusion scintigraphy is more sensitive than multidetector CTPA in detecting chronic thromboembolic pulmonary disease as a treatable cause of pulmonary hypertension. J Nucl Med 2007; 48(5): 680-4.
5. Galiè N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, et al. European Society of Cardiology. Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) andthe European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009; 30(20): 2493-537. Disponible en: http://www.escardio.org/guidelines-surveys/esc-guidelines/GuidelinesDocuments/guidelines-PH-FT.pdf[Consulta: 28/8/09].
6. Jamieson SW. Pulmonary thromboendarterectomy. Heart 1998; 79(2): 118-20.
7. Jamieson SW, Kapelanski DP, Sakakibara N, Manecke GR, Thistlethwaite PA, Kerr KM, et al. Pulmonary endarterectomy: experience and lessons learned in 1,500 cases. Ann Thorac Surg 2003; 76(5): 1457-62.
8. Fedullo PF, Auger WR, Kerr KM, Rubin LJ. Chronic thromboembolic pulmonary hypertension. N Engl J Med 2001; 345(20): 1465-72.
9. Pengo V, Lensing AW, Prins MH, Marchiori A, Davidson BL, Tiozzo F, et al. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med 2004; 350(22): 2257-64.
10. Bonderman D, Skoro-Sajer N, Jakowitsch J, Adlbrecht C, Dunkler D, Taghavi S, et al. Predictors of outcome in chronic thromboembolic pulmonary hypertension. Circulation 2007; 115(16): 2153-8.
11. Bonderman D, Wilkens H, Wakounig S, Schäfers HJ, Jansa P, Lindner J, et al. Lang Risk factors for chronic thromboembolic pulmonary hypertension. Eur Respir J 2009; 33(2): 325-31.
12. Hoeper MM, Mayer E, Simonneau G, Rubin LJ. Chronic thromboembolic pulmonary hypertension. Circulation 2006; 113(16): 2011-20.
13. Bonderman D, Jakowitsch J, Redwan B, Bergmeister H, Renner MK, Panzenböck H, et al. Role for staphylococci in misguided thrombus resolution of chronic thromboembolic pulmonary hypertension. Arterioscler Thromb Vasc Biol 2008; 28(4): 678-84.
14. Egermayer P, Peacock AJ. Is pulmonary embolism a common cause of chronic pulmonary hypertension? Limitations of the embolic hypothesis. Eur Respir J 2000; 15(3): 440-8.
15. Moser KM, Braunwald NS. Successful surgical intervention in severe chronic thromboembolic pulmonary hypertension. Chest 1973; 64(1): 29-35.
16. Lang IM, Klepetko W. Actualización sobre la hipertensión pulmonar tromboembólica crónica, una enfermedad que a menudo no se detecta. Rev Esp Cardiol 2009; 62(2): 120-5.
17. Keogh AM, Mayer E, Benza RL, Corris P, Dartevelle PG, Frost AE, et al. Interventional and surgical modalities of treatment in pulmonary hypertension. J Am Coll Cardiol 2009; 54(1 Suppl): S67-77.
18. Lorente DS, Macchiarini P. Tratamiento quirúrgico en pacientes con hipertensión pulmonar tromboembólica crónica. Arch Bronconeumol 2009; 45(Supl 6): 30-4.
19. Corsico AG, D'Armini AM, Cerveri I, Klersy C, Ansaldo E, Niniano R, et al. Long-term outcome after pulmonary endarterectomy. Am J Respir Crit Care Med 2008; 178(4): 419-24.
20. Kim NH, Fesler P, Channick RN, Knowlton KU, Ben-Yehuda O, Lee SH, et al. Preoperative partitioning of pulmonary vascular resistance correlates with early outcome after thromboendarterectomy for chronic thromboembolic pulmonary hypertension. Circulation 2004; 109(1): 18-22.
21. Dartevelle P, Fadel E, Mussot S, Chapelier A, Hervé P, de Perrot M, et al. Chronic thromboembolic pulmonary hypertension. Eur Respir J 2004; 23(4): 637-48.
22. Jais X, D'Armini AM, Jansa P, Torbicki A, Delcroix M, Ghofrani HA, et al. Bosentan for treatment of inoperable chronic thromboembolic pulmonary hypertension: BENEFiT (Bosentan Effects in iNopErable Forms of chronic Thromboembolic pulmonary hypertension), a randomized, placebo controlled trial. J Am Coll Cardiol 2008; 52(25): 2127-34.
23. Suntharalingam J, Treacy CM, Doughty NJ, Goldsmith K, Soon E, Toshner MR, et al. Long-term use of sildenafil in inoperable chronic thromboembolic pulmonary hypertension. Chest 2008; 134(2): 229-36.
24. Bhatia S, Frantz RP, Severson CJ, Durst LA, McGoon MD. Immediate and long-term hemodynamic and clinical effects of sildenafil in patients with pulmonary arterial hypertension receiving vasodilator therapy. Mayo Clin Proc 2003; 78(10): 1207-13.
25. Michelakis ED, Tymchak W, Noga M, Webster L, Wu XC, Lien D, et al. Long-term treatment with oral sildenafil is safe and improves functional capacity and hemodynamics in patients with pulmonary arterial hypertension. Circulation 2003; 108(17): 2066-9.
26. Ghofrani HA, Schermuly RT, Rose F, Wiedemann R, Kohstall MG, Kreckel A, et al. Sildenafil for long-term treatment of nonoperable chronic thromboembolic pulmonary hypertension. Am J Respir Crit Care Med 2003; 167(8): 1139-41.
27. Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension: the Primary Pulmonary Hypertension Study Group. N Engl J Med 1996; 334(5): 296-302.
28.. Cabrol S, Souza R, Jais X, Fadel E, Ali RH, Humbert M,et al. Intravenous epoprostenol in inoperable chronic thromboembolic pulmonary hypertension. J Heart Lung Transplant 2007; 26(4): 357-62.
29. Olschewski H, Simonneau G, Galiè N, Higenbottam T, Naeije R, Rubin LJ, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med 2002; 347(5): 322-9.
30.. Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002; 346(12): 896-903.
31. Lewczuk J, Piszko P, Jagas J, Porada A, Wójciak S, Sobkowicz B, et al. Prognostic factors in medically treated patients with chronic pulmonary embolism. Chest 2001; 119(3): 818-23.
32. McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation 2006; 114(13): 1417-31.


{kind=link}
{kind=link}