










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkRevista Médica del Uruguay
versión On-line ISSN 1688-0390
Rev. Méd. Urug. vol.25 no.3 Montevideo set. 2009
Policondritis recidivante: presentación clínica, diagnóstico y tratamiento
Dres. Ricardo Silvariño*, María Eugenia Vola†, Patricia Schimchak‡, Ernesto Cairoli§, Juan Alonso¶
Resumen
La policondritis recidivante (PR) es una infrecuente afección autoinmune sistémica, de etiología desconocida y curso recurrente, con mayor frecuencia entre la tercera y cuarta década de la vida. Presentamos un caso de policondritis recidivante, realizando consideraciones clínico-terapéuticas sobre una patología de diagnóstico esencialmente clínico, que se manifiesta con signos y síntomas propios de una enfermedad sistémica, destacándose la necesidad del abordaje multidisciplinario del paciente con enfermedad sistémica inmunomediada.
Palabras clave: POLICONDRITIS RECURRENTE.
Keywords: POLYCHONDRITIS, RELAPSING.
* Asistente Clínica Médica C. Unidad de Enfermedades Autoinmunes Sistémicas. Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay.
† Residente de Oftalmología, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay.
‡ Profesor Adjunto de Oftalmología, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay.
§ Profesor Adjunto Clínica Médica C. Unidad de Enfermedades Autoinmunes Sistémicas. Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Profesor Adjunto del Departamento Básico de Medicina, Hopistal de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay.
¶ Profesor Agregado Clínica Médica C, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay.
Correspondencia: Dr. Ricardo Silvariño
Clínica Médica C, Av Italia s/n y Las Heras, Piso 8. Montevideo, Uruguay
Correo electrónico: rsilvarino@gmail.com
Recibido: 8/6/09.
Aceptado: 27/7/09.
Introducción
La policondritis recidivante (PR) es una afección de origen autoinmune y de curso recurrente, caracterizada por la inflamación crónica y multisistémica del tejido cartilaginoso. Se trata de una entidad poco frecuente, con una incidencia anual estimada de 3,5 casos por millón de habitantes(1). Puede presentarse a cualquier edad, siendo más frecuente entre la tercera y cuarta década de la vida(2). Si bien la etiología es aún desconocida, la presencia de autoanticuerpos contra el colágeno tipo II durante los ataques agudos (proteína presente en el tejido cartilaginoso) y el hallazgo de complejos inmunes circulantes orientan al carácter inmunomediado de su patogenia(3). La primera descripción de la enfermedad data de 1923 en donde fue denominada por Wartenhorst policondropatía(4). En 1960 Pearson(5) introduce el término policondritis y destaca su naturaleza episódica. Mc Adam y colaboradores(6) establecen en 1976 criterios diagnósticos que se exponen en la tabla 1. Los episodios inflamatorios pueden durar días o semanas y se resuelven con tratamiento o de forma espontánea(7). Los cartílagos elásticos de oídos y nariz son los más frecuentemente afectados, aunque otros cartílagos como los articulares periféricos, fibrocartílagos de articulaciones axiales y cartílagos traqueobronquiales pueden verse comprometidos(2).
.
Caso clínico
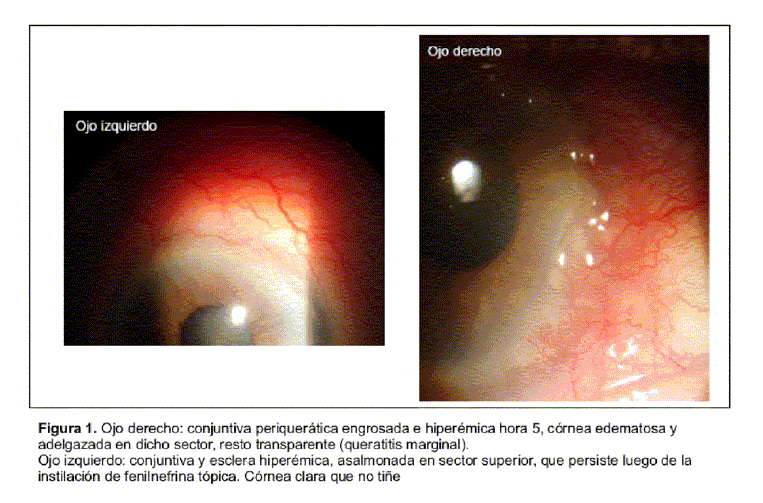
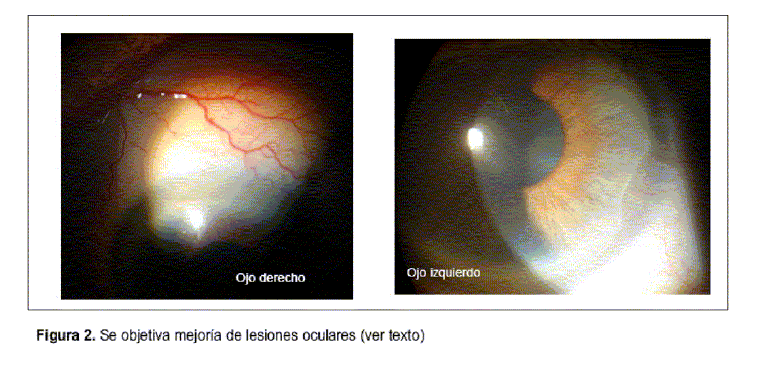
Paciente de 86 años, con antecedentes de diabetes mellitus tipo 2 de reciente diagnóstico, en tratamiento con dieta e hipoglucemiantes orales con adecuado control de glicemia. Presenta historia de 18 meses de evolución, caracterizada por dolor e inflamación del puente nasal, de carácter recurrente, que cede con la ingesta de antinflamatorios no esteroideos. De igual evolución refería episodios de eritema, tumefacción y dolor a nivel de cartílagos de ambos pabellones auriculares, con mayor intensidad a izquierda, que afectaban el helix y ante-helix, respetando lóbulos y sectores no cartilaginosos de ambos pabellones auriculares. Artralgias de pequeñas y medianas articulaciones sin signos fluxivos articulares ni envaramiento matinal. Consultó en el servicio de oftalmología por dolor y enrojecimiento en ojo izquierdo de un mes de evolución, interpretados como blefaroconjuntivitis, realizándose tratamiento tópico sin mejoría ostensible. La paciente reconsulta a la semana observándose en dicha oportunidad en su ojo derecho una conjuntiva hiperémica y engrosada en hora 5 con edema corneal adyacente, en su ojo izquierdo presenta una conjuntiva hiperémica y asalmonada en sector superior que no mejora con la instilación de fenilefrina tópica; en cámara anterior Tyndall y flare positivo (figura 1). El fondo de ojo mostró sectores de atrofia corioretiniana difusa, compatible con miopía elevada sin otras lesiones. Los hallazgos fueron compatibles con escleritis anterior en ojo izquierdo y queratitis periférica en ojo derecho. Del resto del examen físico destacaba la presencia de dolor, rubor y calor en sectores superiores de ambos pabellones auriculares, no detectando afección clínica del cartílago nasal. En el resto del examen osteoarticular, no fueron encontradas alteraciones articulares ni cartilaginosas. El examen cardiovascular y pleuropulmonar fue normal. Con planteo de escleritis anterior se inició tratamiento con corticoides tópicos y lágrimas artificiales obteniendo una mejoría parcial. Se solicitaron estudios en busca de una eventual etiología sistémica responsable de la escleritis destacando: hemograma, examen de orina, azoemia y creatininemia normales, serologías para virus de inmunodeficiencia humana (VIH), serología para lúes (VDRL) y citomegalovirus (CMV) no reactivas, intra-dermorreacción de Mantoux con PPD de 12 mm con baciloscopías negativas y radiografía de tórax sin imágenes patológicas. Se solicitó panel de autoanticuerpos destacándose anticuerpos antinucleares (ANA) y anticuerpos anticitoplasma de neutrófilo (ANCA) negativos, fracciones C3 y C4 del complemento normales y velocidad de sedimentación globular de 20 mm en la primera hora. Dada la coexistencia de condritis nasal y de ambos pabellones auriculares con la característica de ser recurrentes, junto a la afectación ocular con escleritis, se planteó el diagnóstico clínico de policondritis recidivante iniciando tratamiento con prednisona 30 mg vía ora día. Se asoció calcio, vitamina D3 y alendronato para disminuir desmineralización ósea e isoniazida 300 mg vía oral día (durante seis meses) como prevención primaria de infección tuberculosa en una paciente que recibirá corticoides durante un período de tiempo prolongado. Por tratarse de una diabética en quien la terapia corticoidea puede determinar descontrol metabólico se inició metotrexate 10 mg/semana como ahorrador de corticoides. Presentó una buena evolución clínica con remisión del compromiso inflamatorio auricular y mejoría franca de la escleritis (figura 2).
Discusión y comentarios
Entre las manifestaciones más comunes de la PR se encuentran la condritis auricular, condritis nasal, artralgias, artritis no erosiva, síntomas oculares y en menor frecuencia síntomas costocondrales(8,9). En la paciente presentada el debut fue con condritis auricular. La condritis nasal es frecuente, presentándose (20%) con deformación posterior del puente nasal, con nariz en "silla de montar". El antecedente de condritis nasal estuvo presente en la paciente, no constatándose al momento del examen deformaciones del puente nasal. El compromiso articular, presente hasta en 75% en algún momento de la evolución, es habitualmente oligoarticular, no deformante, asimétrico y seronegativo. La presencia de artritis periférica traduce para algunos autores enfermedad diseminada y ensombrece el pronóstico(10). Las artralgias estuvieron presentes en la historia clínica relatada, destacándose la ausencia de artritis y deformaciones articulares. Las manifestaciones oculares son frecuentes y están presentes en 50%-60% de los pacientes(11). Pueden observarse epiescleritis focal o difusa, escleritis anterior y posterior, y más raramente queratitis, iridociclitis, corioretinitis, vasculitis y hemorragias retinianas. La escleritis anterior difusa es el hallazgo más frecuente(12). En una serie recientemente descrita por Tenorio y colaboradores(13) las manifestaciones oculares más frecuentemente halladas fueron epiescleritis, escleritis anterior difusa, iritis y queratitis. La escleritis fue la manifestación que llevó la paciente a consultar, constituyendo en este caso el síntoma y signo clínico que motivó la búsqueda de las otras manifestaciones sistémicas que permitieron arribar al diagnóstico. Hasta 25% de las PR se asocian a otras enfermedades autoinmunes sistémicas (EAS) como poliarteritis nodosa, lupus eritematoso sistémico, artritis reumatoide, enfermedad mixta del tejido conectivo y enfermedad de Sjögren(14). Estas deben ser descartadas frente al diagnóstico de PR. Mención particular entre los diagnósticos diferenciales merece la granulomatosis de Wegener, ya que afecta estructuras oculares y otorrinolaringológicas de forma similar a la PR. La ausencia de manifestaciones clínicas compatibles asociada a la negatividad de los autoanticuerpos solicitados alejan por el momento el planteo de otra enfermedad autoinmune sistémica vinculada a la PR. Dentro de las enfermedades malignas, los linfomas (Hodgkin y no Hodgkin), la leucemia mielomonocítica crónica y algunos carcinomas sólidos pueden vincularse a su desarrollo, e incluso preceder en el tiempo a la PR(15). No hallamos evidencia clínica de una neoplasia actual. El diagnóstico es eminentemente clínico, aunque muchas situaciones pueden simular manifestaciones de la PR. En quienes la presentación clínica es característica no es necesaria la biopsia para confirmar el diagnóstico(16). La presencia de tres de los criterios diagnósticos sugeridos por Mc Adam(6) permitieron plantear el diagnóstico de PR, pudiendo prescindir de la biopsia frente a dicho cuadro clínico. Destacamos que los criterios diagnóstico sugeridos fueron formulados sobre una base empírica, basados en experiencia y series personales, sin grupos control y careciendo de estimaciones de especificidad, sensibilidad y valor predictivo negativo y positivo. Los hallazgos de laboratorio son inespecíficos con aumento de la velocidad de sedimentación globular, anemia de perfil inflamatorio, leucocitosis, trombocitosis e hipergamaglobulinemia policlonal(16), no presentes en la paciente. Cuando coexiste con otra enfermedad autoinmune sistémica puede observarse positividad de los ANA y factor reumatoideo (FR)(17,18), siendo negativos en esta oportunidad. Cuando existe afectación renal se han descrito casos con positividad de ANCA(19), no presentes en la paciente. Hasta en 50% de los pacientes pueden hallarse anticuerpos anticolágeno tipo II, sugiriéndose que el nivel de los mismos podría correlacionarse con la actividad de la enfermedad(20). No contamos en nuestro medio con esta herramienta diagnóstica. El tratamiento cuando la manifestación es la condritis es en base a antiinflamatorios no esteroideos (AINE) y dosis bajas de prednisona vía oral. Cuando están presentes manifestaciones laringotraqueales, oculares o glomerulonefritis se inicia prednisona a dosis de 1 mg/k/día. Una vez obtenida respuesta se comienza el descenso gradual de los corticoides hasta su retirada. Pueden requerirse dosis bajas de prednisona como mantenimiento si se observan recurrencias frente a la suspensión de los corticoides. Cuando existe refractariedad o las dosis de corticoides para mantener al paciente libre de síntomas son elevadas pueden utilizarse inmunosupresores como ahorradores de corticoides tales como metotrexate o azatioprina(21).
En la paciente presentada se objetivó buena respuesta al tratamiento corticoideo. La introducción precoz de un ahorrador de corticoides se fundamentó en la comorbilidad que presentaba. La sobrevida en la primera serie publicada por Mc Adam(6) en 1976 fue 70% a cuatro años. Michet(22) reporta en 1986 una sobrevida de 74% a cinco años. En la serie publicada por Trentham(2) en 1998 la supervivencia a 8 años fue de 94%, lo que puede adjudicarse a los progresos en el tratamiento de la enfermedad. Entre los factores que predijeron disminución de la sobrevida en la cohorte de Michet se citan edad avanzada, presencia de anemia al momento del diagnóstico y estenosis laringotraqueal. La paciente presentada tenía edad avanzada al momento del diagnóstico como único factor predictor de mala evolución.
Conclusiones
La PR es una enfermedad poco frecuente y de diagnóstico esencialmente clínico, por lo que su conocimiento asociado a la sospecha clínica son fundamentales para establecer su diagnóstico. Al igual que otras EAS se manifiesta con signos y síntomas en cualquier sector de la economía, lo que remarca la necesidad del abordaje multidisciplinario del paciente con enfermedad sistémica inmunomediada. Dado que algunas manifestaciones clínicas (principalmente la escleritis) pueden responder a infecciones específicas, éstas deben ser descartadas durante el proceso diagnóstico. La coexistencia de PR en el contexto de otras EAS hace razonable la búsqueda de las mismas por la clínica y el laboratorio.
Summary
Relapsing polychondritis (RP) is an unusual systemic autoimmune disease, of unknown etiology and recurrent nature, that is more frequent between the third and the fourth decade of life. We present a case of relapsing polychondritis, stating clinical and therapeutic issues about a disease that is clinically diagnosed, with the typical signs and symptoms of a systemic disease, and emphasize on the need for a multidisciplinary approach of the patient suffering from an immune-mediates systemic disease.
Résumé
La polychondrite récidivante est une maladie auto-immune peu fréquente, systémique, à étiologie inconnue et récurrente qui se présente plus fréquemment pendant la troisième et la quatrième décade de la vie. On présente ici un cas de polychondrite récidivante dont on fait des remarques cliniques et thérapeutiques; il s’agit d’une pathologie à diagnostic essentiellement clinique ayant les symptômes et les signes d’une maladie systémique. On met l’accent sur l’importance d’un abord multidisciplinaire du patient.
Resumo
A policondrite recidivante (PR) é uma afecção auto-imune sistêmica pouco frequente, de etiologia desconhecida e recorrente, com maior frequencia nas terceira e quarta década de vida. Apresentamos um caso de policondrite recidivante, realizando considerações clínico-terapêuticas sobre una patologia de diagnóstico essencialmente clínico, que se manifesta com sinais e sintomas próprios de uma doença sistêmica, ressaltando a necessidade da abordagem multiprofissional do paciente com doença sistêmica com componente imunológico.
Bibliografía
1. Zeuner M, Straub RH, Rauh G, Albert ED, Schölmerich J, Lang B. Relapsing polychondritis: clinical and immunogenetic analysis of 62 patients. J Rheumatol 1997; 24(1): 96-101.
2. Trentham DE, Le CH. Relapsing polychondritis. Ann Intern Med 1998; 129(2): 114-22.
3. Letko E, Zafiraskis P, BaltaTzis S, Voudouri A, Livir-Rallatos C, Foster CS. Relapsing polychondritis: a clinical review. Semin Arthritis Rheum 2002; 31(6): 384-95.
4. Jaksch-Wartenhorst R. Polychondropathia. Wien Arch Intern Med 1923; 6: 93-100.
5. Pearson CM, Kline HM, Newcomer VD. Relapsing polychondritis. N Engl J Med 1960; 263: 51-8.
6. McAdam LP, O’Hanlan MA, Bluestone R, Pearson CM. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore) 1976; 55(3): 193-215.
7. Luthra HS, Michet CJ. Relapsing polychondritis. En: Klippel JH, Dieppe PA. Rheumathology. St Louis: Mosby, 1994: 31.1-31.4.
8. Meza J, Remes J, Montaño A. Policondritis recidivante: presentación de 5 casos y revisión de la literatura. Rev Mex Reumatol 2001; 16(5): 309-14.
9. Díaz A, Dolinsky D, Prodanov A, Schiavo L, Liñares L. Policondritis Recidivante. Cuad Med Interna (Uruguay) 2002: 52-6.
10. Balsa A, Expinosa A, Cuesta M, MacLeod TI, Gijón-Baños J, Maddison PJ.
11. Chow MT, Anderson SF. Relapsing polychondritis. Optom Vis Sci 2000; 77(6): 286-92.
12. Hoang-Xaun T, Foster CS, Rice BA. Scleritis in relapsing polychondritis. Resonse to therapy. Ophthalmology 1990; 97(7): 892-8.
13. Tenorio G, Bustos R, Lino L. Manifestaciones sistémicas y oculares de la policondritis recurrente. Rev Med Hosp Gen Mex 2004; 67(4): 189-95.
14. Fariña M, Fernández A, Consani S, Cuadro R, Larre Borges A. Asociación de nefropatía por Sindrome de Sjogren y Policondritis Recidivante. Arch Med Interna (Montevideo) 2008; 30(2-3): 53-78.
15. Firestein GS, Gruber HE, Weisman MH, Zvaifler NJ, Barber J, O’Duffy JD. Mouth and genital ulcers with inflamed cartilage: MAGIC syndrome. Five patients with features of relapsing polychondritis and Behçet’s disease. Am J Med 1985; 79(1): 65-72.
16. Hochberg M. Policondritis Recidivante. En: Ruddy S, Harris E, Sledge C, Budd R, Sergent J. Kelley´s reumatología. 6ª ed. Madrid: Marbán, 2003: 1463-7.
17. Franssen MJ, Boerbooms AM, van de Putte LB. Polychondritis and rheumatoid artritis: Case report and review of the literature. Clin Rheumatol 1987; 6(3): 453-7.
18. Harisdangkul V, Johnson WW. Association between relapsing polychondritis and systemic lupus erythematosus. South Med J 1994; 87(7): 753-7.
19. Papo T, Piette JC, Le Thi Huong D, Godeau P, Meyer O, Kahn MF, et al. Antineutrophil cytoplasmic antibodies in polychondritis. Ann Rheum Dis 1993; 52(5): 384-5.
20. Ebringer R, Rook G, Swana GT, Bottazzo GF, Doniach D. Autoantibodies to cartilage and tipe II collagen in relapsing polychondritis and other rheumatic diseases. Ann Rheum Dis 1981; 40(5): 473-9.
21. Jabs DA, Rosenbaum JT, Foster CS, Holland GN, Jaffe GJ, Louie JS, et al. Guidelines for the use of immunosuppressive drugs in patients with ocular inflammatory disorders: recommendations of an expert panel. Am J Ophthalmol 2000; 130(4): 492-513.
22. Michet CJ Jr, McKenna CH, Luthra HS, O’Fallon WM. Relapsing polichondrytis: survival and predictive role of early disease manifestations. Ann Intern Med 1986; 104(1): 74-8.


{kind=link}
{kind=link}
{kind=link}