










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkRevista Médica del Uruguay
On-line version ISSN 1688-0390
Rev. Méd. Urug. vol.25 no.2 Montevideo June 2009
Manifestaciones clínicas en pacientes con esclerosis sistémica
Ricardo Silvariño*, Martín Rebella*, Juan Alonso†, Ernesto Cairoli‡
Unidad de Enfermedades Autoinmunes Sistémicas, Clínica Médica "C", Departamento Clínico de Medicina, Hospital de Clínicas, Universidad de la República. Montevideo, Uruguay
Resumen
Introducción: la esclerosis sistémica es una enfermedad autoinmune sistémica caracterizada por daño endotelial, inflamación y fibrosis de piel, vasos y órganos internos. Existen diferencias en frecuencia, gravedad y pronóstico en diferentes grupos étnicos, lo que remarca la importancia del estudio en cada región geográfica, para poder diagnosticar tempranamente sus manifestaciones incipientes.
Material y método: realizamos un estudio descriptivo y retrospectivo entre marzo de 2006 y marzo de 2008, incluyendo pacientes con diagnóstico definitivo de esclerosis sistémica, asistidos en la Unidad de Enfermedades Autoinmunes Sistémicas del Hospital de Clínicas.
Resultados: fueron incluidas 31 mujeres, con 39,2 meses de seguimiento promedio, edad media al diagnóstico de 47,6 años. Once pacientes (35,5%) presentaron enfermedad difusa y 20 (64,5%) enfermedad limitada. Treinta presentaron fenómeno de Raynaud. En 92% la capilaroscopia mostró un patrón esclerodermiforme. A nivel respiratorio encontramos intersticiopatía en 25%, hipertensión arterial pulmonar en 22,2% y un patrón restrictivo en estudios de función respiratoria en 35,5%. El 67,7% presentó manifestaciones digestivas y 9,6% desarrolló una crisis renal esclerodérmica. Hallamos anticuerpos antinucleares (ANA) en 29/31 (93,5%) pacientes; 16 presentaron anticuerpos anticentromero y cinco anti-topoisomerasa-I. Las cuatro (12,9%) pacientes fallecidas durante el seguimiento presentaron elementos comunes como esclerosis sistémica difusa, úlceras digitales y compromiso respiratorio severo.
Conclusiones: las características clínicas e inmunológicas encontradas fueron similares a las descriptas en otras series. En ausencia de tratamiento específico, debe insistirse en la valoración periódica de las repercusiones viscerales para controlar y retrasar la instalación de complicaciones con elevada morbimortalidad.
Palabras clave: ESCLERODERMIA SISTÉMICA.
Keywords: SCLERODERMA, SYSTEMIC.
* Asistente de Clínica Médica, Clínica Medica "C", Departamento Clínico de Medicina, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay.
† Profesor Agregado de Clínica Médica, Clínica Medica "C", Departamento Clínico de Medicina, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay.
‡ Profesor Adjunto de Clínica Médica, Profesor Adjunto del Departamento Básico de Medicina. Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay.
Correspondencia: Dr. Ernesto Cairoli
Unidad de Enfermedades Autoinmunes Sistémicas, Clínica Médica "C", Departamento Clínico de Medicina, Hospital de Clínicas,
Universidad de la República.
Av. Italia s/n piso 8. Montevideo, Uruguay
Correo electrónico: ecairoli@hc.edu.uy
Recibido: 8/12/08.
Aceptado: 23/3/09.
Introducción
La esclerosis sistémica (ES) (también denominada esclerodermia) es una enfermedad autoinmune sistémica caracterizada por la inflamación y fibrosis principalmente cutánea, concomitante al daño endotelial y afección inflamatoria de vasos sanguíneos, tejidos osteomusculares y órganos internos. Según la extensión del compromiso cutáneo se puede clasificar la ES en difusa (ESD) o ES limitada (ESL). La primera se caracteriza por el endurecimiento generalizado de la piel, compromiso frecuente de órganos internos y presencia de anticuerpos antitopoisomerasa I (Scl-70), siendo esta presentación clínica la de peor pronóstico. La ESL se caracteriza por el endurecimiento de la piel a nivel distal en extremidades, cara y cuello, menor compromiso de órganos internos, presencia de anticuerpos anticentrómero y mejor pronóstico comparada con la ESD. Otra variante de menor frecuencia es la ES sin esclerodermia, donde existe afectación de órganos internos sin compromiso de piel(1). Existe una gran variabilidad en las manifestaciones clínicas y biológicas de la enfermedad. Se han reportado diferencias en frecuencia, gravedad de las manifestaciones clínicas y pronóstico en los diferentes grupos étnicos estudiados. Esto marca la importancia del estudio de las características clínicas y biológicas de la enfermedad en cada región, analizando la presencia de variables genéticas y ambientales y su incidencia en la presentación clínica y pronóstica. Conocer las diferentes modalidades de presentación clínica de la ES en nuestro medio permitiría tener la sospecha precoz, establecer un diagnóstico temprano y poder anticiparnos a la aparición de repercusiones severas, utilizando el uso sistemático de estudios paraclínicos, evitando la instalación de secuelas que incrementarían la morbimortalidad de estos pacientes.
Revisión de la literatura
La sobreproducción de matriz extracelular por parte de los fibroblastos es el principal marcador de la esclerodermia. La acumulación de colágeno es resultado de una interacción anormal entre las células endoteliales, las células mononucleares (linfocitos y monocitos) y los fibroblastos, lo que conduce a la producción de citoquinas inductoras de fibrosis, en un contexto de hiperreactividad vascular e hipoxia tisular(2). La fibrosis cutánea es característica de la ES, con buena correlación entre extensión de la esclerosis y sobrevida(3). El fenómeno de Raynaud está presente en más de 90% de pacientes con ES y en 90% de estos está asociado a la presencia de fibrosis de dedos, pérdida de pulpejos, úlceras y amputación digital(4). Acroesclerosis y esclerodactilia son halladas en 100% en algunas series y la presencia de cicatrices en pulpejos hasta en 85%(5). La enfermedad pulmonar es frecuente, presente tanto en la forma limitada como difusa, siendo las complicaciones a este nivel la principal causa de mortalidad(6). La enfermedad intersticial es la manifestación más frecuente, detectada en 40% de pacientes sometidos a pruebas de función respiratoria y en 70%-80% de los estudios postmórtem(7). En las pruebas de función respiratoria (volúmenes pulmonares y espirometría) presentan habitualmente un patrón de tipo restrictivo. Las alteraciones más tempranas se evidencian por una disminución en la difusión del monóxido de carbono (DLco) en las pruebas funcionales(8). La DLco es el mejor índice de extensión del daño pulmonar, mostrándose para tales fines más sensible que la tomografía de alta resolución (TCar)(9). La TCar es el estándar para el diagnóstico de enfermedad del intersticio, evidenciando alteraciones hasta en 44% de los pacientes que cursan con radiografía de tórax normal(10). La hipertensión arterial pulmonar (HTP) puede ser secundaria al compromiso intersticial o presentarse por vasculopatía como puede observarse en la ESL(11). El estudio no invasivo para su detección (de manera indirecta) es la ecocardiografía, siendo el cateterismo cardíaco derecho el que confirma la presencia de HTP. El test de la marcha de seis minutos brinda valiosa información, ya que es predictor de morbilidad y mortalidad en portadores de HTP(11).
La afectación renal es una complicación grave mostrando algunas series hasta 50% de pacientes con hipertensión, proteinuria o hiperazoemia(12). La crisis renal esclerodérmica se caracteriza por hipertensión arterial acelerada o insuficiencia renal oligúrica rápidamente progresiva, o ambas, presente hasta en 20% de pacientes con ESD, pudiendo en menor proporción (menos de 10%) presentar una crisis renal sin hipertensión arterial (crisis renal esclerodérmica normotensiva)(13).
La prevalencia de afectación cardíaca clínica es baja pero evidente mediante valoración por ecocardiografía y otros métodos diagnósticos. La disfunción sistólica ventricular izquierda es a menudo subclínica, habiéndose detectado hasta en 89% de pacientes asintomáticos(14). Es frecuente el hallazgo de disfunción diastólica en el contexto de ESD, en pacientes sin hipertensión arterial sistémica(15). La disfunción ventricular derecha es también frecuente como resultado de la HTP asociada(16), con o sin enfermedad del intersticio pulmonar. Su presencia se vincula a un peor pronóstico vital(17). La prevalencia de enfermedad coronaria aterosclerótica no está incrementada en la ES(18), sin embargo, en aquellos que tienen enfermedad coronaria el vasoespasmo asociado es más frecuente e intenso que en la población general(19).
Entre las alteraciones endocrinológicas, el hipotiroidismo (hasta 43%), la hiperprolactinemia (13%-59%) y la osteopenia-osteoporosis (35%-44%) son patologías frecuentes en el paciente con ES(20).
La afectación gastrointestinal es frecuente. Se detecta disminución de la motilidad esofágica hasta en 90% de los pacientes, con afectación predominante de los dos tercios distales del esófago(21). La enfermedad por reflujo gastroesofágico es una complicación muy frecuente, consecuencia de la disminución de la presión del esfínter esofágico inferior, hipomotilidad esofágica y gastroparesia(22).
La capilaroscopia del lecho ungueal es una herramienta útil en el diagnóstico y seguimiento de la enfermedad(23). Se han propuesto tres nuevos patrones designados como patrón precoz, activo y tardío, que se correlacionan con la duración de la enfermedad y del fenómeno de Raynaud(24).
Los anticuerpos antinucleares (ANA) están presentes en 90%-100% de pacientes con ES. Los Scl-70 están presentes en 40% de los casos y habitualmente en pacientes con la forma difusa, estando asociados al desarrollo de alveolitis fibrosante. Los anticuerpos anticentrómeros están presentes en 70%-80% de los casos con la forma limitada y muestran asociación con el desarrollo de HTP(25).
El objetivo de este trabajo fue determinar las características clínicas de pacientes con ES en seguimiento periódico en una unidad clínica especializada, poniendo especial énfasis en el compromiso clínico cutáneo, visceral e inmunitario.
Material y método
Se realizó un estudio descriptivo y retrospectivo, obteniendo información de los registros médicos de pacientes con ES asistidos en la Unidad de Enfermedades Autoinmunes Sistémicas, Clínica Médica "C" del Departamento Clínico de Medicina, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Fueron incluidos pacientes con diagnóstico definitivo de ES según criterios de clasificación propuestos por el Colegio Americano de Reumatología(26), asistidos en el período comprendido entre marzo de 2006 y marzo de 2008. En función de la distribución de la esclerosis cutánea se los clasificó en formas difusa y limitada. Fueron excluidos pacientes con esclerosis cutánea pero con diagnóstico de síndrome indiferenciado, de superposición y conectivopatía mixta del tejido conectivo. Se elaboró una tabla electrónica para la recolección de datos y posterior análisis estadístico, registrando las siguientes características: edad, sexo, procedencia urbana o rural, nivel de instrucción, compromiso cutáneo, osteoarticular y visceral (respiratorio, digestivo, renal, vascular) así como autoanticuerpos presentes y comorbilidad asociada.
Análisis estadístico
Los resultados se expresaron en valores absolutos, porcentajes, medias y sus respectivos desvíos estándar (± SD). Para la comparación de datos de la muestra (en los casos que correspondió) se utilizaron test no paramétricos (test de Student y Mann-Whitney U test). Se consideraron como significativos valores de p<0,05.
Resultados
Fueron incluidos 31 pacientes, todos de sexo femenino y provenientes de zona urbana. La media de edad al momento de la inclusión en el estudio fue de 50,7 años (± 13,4) (rango 20-70 años). La edad al momento del diagnóstico fue de 47,6 años (± 14,4). La media de seguimiento fue de 39,2 meses (± 34,2). Del total de los incluidos, 11 (35,5%) se presentaron con ESD y 20 (64,5%) con ESL. En cuanto al fenómeno de Raynaud encontramos que 30/31 (96,7%) pacientes lo presentaron en algún momento de la evolución de la enfermedad. La media de duración en el total de la muestra al momento del estudio fue de 129,9 meses (± 99) (rango 19-384 meses). Comparando las medias de duración del fenómeno de Raynaud entre pacientes con ESD y ESL, encontramos una media de 127 meses (± 118) y de 131,5 meses (± 89,7), respectivamente, diferencias estadísticamente no significativas (p=0,302). El tiempo entre el diagnóstico del fenómeno de Raynaud y la realización del diagnóstico de ES fue menor en los casos de enfermedad difusa (60 meses) comparado con la forma limitada (90,7 meses), donde si bien se llegó más tempranamente al diagnóstico de ESD, las diferencias no fueron estadísticamente significativas (p=0,085).
La capilaroscopia periungueal se realizó en 26/31, de los cuales en 24 casos (92,3%) se describió un patrón esclerodermiforme. Desarrollaron úlceras digitales en algún momento de la evolución 12/31 (38,7%) pacientes y 3/31 (9,6%) evolucionaron a la necrosis digital, requiriendo amputación quirúrgica de una falange. En referencia al compromiso de cutáneo, todos los pacientes presentaron esclerosis cutánea con variable grado de extensión y severidad; 11/31 (35,5%) presentaron esclerodactilia, 15/31 (48,3%) microstomía, 14/31 (45,1%) telangiectasias y 3/31 (9,6%) calcinosis.
El compromiso pulmonar se caracterizó en lo funcional por el hallazgo en 11/31 (35,5%) pacientes de un patrón restrictivo evaluado mediante espirometría. Se realizó TCar torácica en 25/31 (80,6%) pacientes, presentando elementos compatibles con enfermedad pulmonar intersticial difusa 25% de los casos.
En 27 pacientes obtuvimos datos de la ecocardiografía, encontrando que seis (22,2%) presentaron signos indirectos de HTP, donde la presión sistólica de arteria pulmonar media fue 60,4 mmhg (± 28,6). De ellos, cuatro presentaban una ESD y dos una ESL. En dos casos la HTP estuvo asociada a enfermedad pulmonar intersticial pulmonar.
En referencia a otros compromisos viscerales, 21/31 (67,7%) presentaron manifestaciones digestivas clínicas o radiológicas, o ambas. El reflujo gastroesofágico fue la manifestación digestiva más frecuente estando presente en 21/31 (67,7%) pacientes. Respecto al compromiso renal, 3/31 (9,6%) presentaron una crisis renal esclerodérmica, manteniendo alterado el filtrado glomerular en forma persistente, ingresando uno de ellos en plan de hemodiálisis crónica.
El factor de riesgo cardiovascular más prevalente fue la hipertensión arterial hallada en 11/31 (35,5%), seguida del tabaquismo presente en 4/31 (12,9%).
El perfil de autoanticuerpos evidenció la presencia de ANA en 29/31 (93,5%). De ellos, 16 (51,6%) tuvieron anticuerpos anticentrómero y cinco (16,1%) Scl-70.
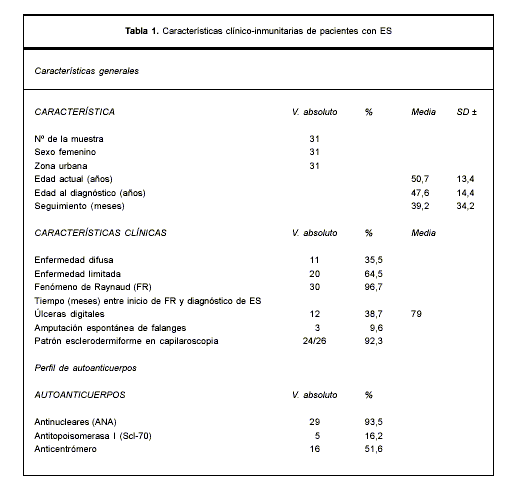
En el período de seguimiento fallecieron cuatro pacientes (12,9%). La media de edad fue de 37,7 años (rango 22-52 años), menor a la hallada como media del total de las pacientes. Todas presentaban ES en su forma difusa, HTP, alteraciones en la capilaroscopia periungueal y ANA positivos. En tres de ellas la causa de la muerte estuvo relacionada con afecciones respiratorias (insuficiencia respiratoria, HTP, infecciones), no disponiendo de datos de la paciente restante. En la tabla 1 se resumen las características clínicas e inmunitarias del grupo de pacientes estudiado.
Discusión
En comparación con la serie publicada por Rojas-Serrano y colaboradores(5), hallamos mayor edad al diagnóstico en nuestra serie (35,6 ± 15 versus 47,6 ± 14,4), similar a lo observado por Simeon y colaboradores(27) (48,8 años) y Quadrelli y colaboradores(28) (47,7 ± 10,2 años). En otras series se incluyeron casos de ES en hombres, contrariamente a nuestros resultados donde el total de pacientes son de sexo femenino. El tiempo entre el inicio del fenómeno de Raynaud y el establecimiento del diagnóstico de ES fue mayor al descripto en la forma difusa y de similar duración para la forma limitada(2). Al igual que Quadrelli y colaboradores, hallamos un elevado número de pacientes con capilaroscopia patológica (77,8% y 92,3%, respectivamente). Encontramos menor incidencia de esclerodactilia (48,3% versus 100%) y positividad de ANA (93,5% versus 100%) comparado con el grupo de Rojas-Serrano. En la serie descripta por Quadrelli, 31,4% presentó esclerodactilia y 90,4% positividad de ANA. Al igual que Simeon y colaboradores (87,7%), hallamos compromiso esofágico en un elevado número de pacientes (67,7%). La frecuencia de crisis renal esclerodérmica fue similar a la hallada en otros grupos donde se reporta en aproximadamente 10% de los casos. La fibrosis pulmonar e HTP fueron observadas con similares frecuencias a las descriptas en la serie de Simeon (12,9% versus 17% y 19,4% versus 17%, respectivamente). Quadrelli encuentra HTP en 15,3% y fibrosis pulmonar en 44,1%. La positividad de autoanticuerpos fue similar a la reportada por Quadrelli (antitopoisomerasa I: 16,2% versus 20% y anticentrómero 51,6% versus 55%).
La mortalidad fue más elevada que la observada por Rojas-Serrano (12,9% versus 5%) en similar período de seguimiento. En este sentido, uno de los factores que inciden en esta diferencia es el tratamiento de la HTP con fármacos específicos, donde algunos de ellos (con distinta eficacia) han demostrado disminuir la morbimortalidad de estos pacientes. Al igual que otros autores(29), observamos que los decesos ocurrieron en pacientes jóvenes, con forma difusa de la ES y con enfermedad rápidamente progresiva, apoyando lo descripto en otras series, donde la mayor extensión del compromiso cutáneo se asoció a mayor mortalidad. El compromiso pulmonar en todas sus modalidades se mostró en nuestra serie como factor común entre las fallecidas en el período de seguimiento, coincidiendo con lo observado por Ioannidis y colaboradores(30), que describen el compromiso pulmonar, renal, cardiovascular y presencia de anticuerpos antitopoisomerasa como predictores de mortalidad. Asimismo, Chung y colaboradores(31) encuentran que la fibrosis pulmonar es un fuerte predictor de mal pronóstico en pacientes con ES que requirieron internación.
La totalidad de las fallecidas mostraron positividad para los ANA, encontrando evidencias(32) que los vinculan a mayor compromiso visceral y reducción de la sobrevida.
El seguimiento longitudinal de nuestra serie fue de 39,2 ± 34 meses, donde algunas de las manifestaciones evolutivas de la enfermedad probablemente hayan quedado excluidas en este período de observación. Al respecto vale destacar que la mayor incidencia de daño grave de órgano blanco ocurrió al igual que en las series citadas en los primeros tres años desde el inicio de los síntomas(5,27,28).
Conclusiones
Exceptuando la media de edad al diagnóstico (mayor en nuestra serie), hallamos similares características clínicas e inmunológicas a las descriptas en otras series consultadas. La mortalidad fue mayor que la descripta por otros autores. La extensión de la fibrosis cutánea, la positividad de los ANA y el compromiso pulmonar fueron características comunes entre las pacientes fallecidas durante el seguimiento. Estas presentaron cuadros clínicos de rápida evolución, por lo que en ausencia de tratamientos inmunosupresores eficaces para detener la progresión de la enfermedad debe insistirse en la valoración periódica y lograr la detección precoz de las repercusiones a nivel cutáneo, digestivo, respiratorio, renal y cardíaco.
Al momento no se dispone de ningún tratamiento inmunosupresor con suficiente nivel de evidencia que demuestre ser capaz de evitar la progresión de la ES. Existen ensayos prospectivos cuyos resultados preliminares muestran al ácido micofenólico (micofenolato de mofetilo y micofenolato sódico), como una alternativa posible en casos de daño respiratorio(33-35).
El tratamiento a implementar será entonces el de las complicaciones, buscando evitar su progresión y el empeoramiento en la calidad de vida. Esto es particularmente importante para los casos de crisis renal esclerodérmica y de HTP. En los primeros, el tratamiento de la hipertensión arterial basado en el uso de inhibidores de enzima conversora de angiotensina (IECA) solos o asociados a los antagonistas del receptor de angiotensina II (ARA II) lograron disminuir los casos de fallo renal y muerte. En el caso de la HTP, su detección precoz (mediante ecocardiografía y estudio funcional respiratorio con DLco) y el inicio terapéutico de anticoagulantes orales (warfarina) simultáneamente con antagonistas de los canales de calcio (diltiazem), inhibidores de la 5 fosfo-diesterasa (sildenafil), antagonistas del receptor de la endotelina (bosentan) o de análogos de la prostaciclina (epoprostenol, iloprost) han demostrado en todos los casos mejorar la calidad de vida, mejoras en el test de la marcha a seis minutos y en algunos de ellos una reducción significativa de la mortalidad(34).
Summary
Introduction: systemic sclerosis is an autoimmune disease characterized by endothelial damage, and skin, vessel and internal organ fibrosis and inflammation. There are differences in terms of frequency, severity and prognosis for the different ethnic groups, what reinforces the importance of the study in each geographical region with the purpose of enabling early diagnosis of its incipient symptoms.
Methods: we conducted a descriptive and retrospective study form March 2006 through March 2008, including patients with a final diagnosis of systemic sclerosis, who are treated at the Systemic Autoimmune Diseases Unit at the Clínicas Hospital.
Results: 31 women were included in the study, average follow-up of patients was 39.2 months, and average age at the time of diagnosis was 47.6 years. Eleven patients (35,5%) presented diffuse disease and 20 (64.5%) of them evidenced limited disease. Thirty patients presented Raynaud’s phenomenon. In 92% of cases capilaroscopy showed a sclerodermiform pattern. In terms of the respiratory system, we found interstilial pathology in 25% of cases, pulmonary arterial hypertension in 22.2 % and a restrictive pattern in respiratory function studies in 35.5%. Also, 67.7% presented digestive manifestations and 9.6% developed sclerodermic renal crisis. We found anti-nuclear antibodies (ANA) in 29 out of 31 patients (93,5%) patients; 16 presented anticentromere antibodies and five anti-topoisomerasa-I antibodies. The four patients (12.9%) who died during follow-up presented common elements such as diffuse sclerosis, digital ulcers and severe respiratory compromise.
Conclusions: the clinical and immune characteristics found in our study were similar to those described in other series. Should there be no specific treatment, it is essential to perform regular assessment of visceral impact in order to control and delay complications which result in high morbimortality rates.
Résumé
Introduction: la sclérose systémique est une maladie auto immune systémique caractérisée par des lésions de la peau, inflammation et progression de tissu fibreux, capillaires sanguins et organes internes. Il existe des différences en ce qui concerne la fréquence, la gravité et le pronostic aux différents groupes ethniques, ce qui souligne l’importance son étude à chaque région géographique pour en faire le diagnostic précoce.
Matériel et méthode: on fait ici une étude descriptive et rétrospective de mars 2006 à mars 2008, qui inclut des patients à diagnostic définitif de sclérose systémique, assistés à l’Unité de Maladies Auto immunes Systémiques de l’Hôpital de Clinicas.
Résultats: on inclut 31 femmes qui avaient été objet d’ un suivi moyen de 39,2 mois, âge moyenne 47,6 ans au moment du diagnostic. Onze patientes (35,5%) ont présenté maladie diffuse et 20 (64,5%) maladie limitée. Trente ont présenté phénomène de Raynaud. Chez 92%, la capillaroscopie a montré un patron scléro-dermiforme. Au niveau respiratoire, on trouve intersticiopathie chez 25%, hypertension artérielle pulmonaire chez 22,2% et une constante restrictive aux études de fonction respiratoire chez 35,5% des femmes. 67,7% a eu des manifestations digesti-ves et 9,6% une crise rénale scléro-dermique. On a trouvé des anticorps anti-nucléaires (ANA) à 29/31 (93,5%) patientes; 16 ont présenté des anticorps anti-centromère et cinq anti-topoisomérase-I. Les quatre, 12,9%, patientes décédées pendant le suivi, ont présenté des éléments communs tels que sclérose systémique diffuse, ulcères digitales et trouble respiratoire sévère.
Conclusions: les caractéristiques cliniques et immuno-logiques trouvées ont été semblables à celles décrites dans d’autres séries. Face à l’absence de traitement spécifique, on doit insister sur le suivi périodique des répercussions viscérales afin de contrôler et de retarder l’installation des troubles à haute morbi-mortalité.
Resumo
Introdução: a esclerose sistêmica é uma doença auto-imune sistêmica caracterizada por dano endotelial, infla-mação e fibrose de pele, vasos e órgãos internos. Existem diferenças na freqüência, severidade e prognóstico nos diferentes grupos étnicos, por isso é importante estudá-la em diferentes regiões geográficas para o diagnóstico precoce de suas manifestações.
Material e método: realizamos um estudo descritivo e retrospectivo entre março de 2006 e março de 2008, incluindo pacientes com diagnóstico definitivo de esclerose sistêmica, atendidos na Unidade de Doenças Auto-imunes Sistêmicas do Hospital das Clínicas.
Resultados: foram incluídas 31 mulheres, com um seguimento médio de 39,2 meses, idade média no diagnóstico de 47,6 anos. Onze pacientes (35,5%) apresentaram doença difusa e 20 (64,5%) doença limitada. Trinta apresentaram fenômeno de Raynaud. Em 92% a capilaroscopia mostrou um padrão esclerodermiforme. No aparelho respiratório encontramos intersticiopatia em 25%, hipertensão arterial pulmonar em 22,2% e um padrão restritivo nos estudos de função respiratória em 35,5%. 67,7% apresentaram manifestações digestivas e 9,6% crise renal esclerodérmica. Em 29/31 (93,5%) pacientes encontramos anticorpos antinucleares (ANA); 16 apresentaram anticorpos anticentrômero e cinco anti-topoisomerase-I. As quatro (12,9%) pacientes que faleceram durante o seguimento apresentaram elementos comuns como esclerose sistêmica difusa, úlceras digitais e compromisso respiratório severo.
Conclusões: as características clínicas e imunológicas encontradas foram similares às descritas em outras séries. Na ausência de tratamento específico, deve-se insistir na avaliação periódica das repercussões viscerais para controlar e retardar a instalação de complicações com morbimortalidade elevada.
Bibliografía
1. LeRoy EC. Systemic sclerosis. A vascular perspective. Rheum Dis Clin North Am 1996; 22(4): 675-94.
2. Roa Johanna. Avances en la clasificación, inmunopatogenia y tratamiento de la esclerodermia. Reumatología 2005; 21(1): 27-32.
3. Shand L, Lunt M, Nihtyanova S, Hoseini M, Silman A, Black CM, et al. Relationship between change in skin score and disease outcome in diffuse cutaneous systemic sclerosis: application of a latent linear trajectory model. Arthritis Rheum 2007; 56(7): 2422-31.
4. Flavahan NA, Flavahan S, Mitra S, Chotani MA. The vasculopathy of Raynaud’s phenomenon and scleroderma. Rheum Dis Clin North Am 2003; 29(2): 275-91.
5. Rojas-Serrano J, Codina H, Medrano G, Abraham J, Vera O, Vázquez J. Menor incidencia de daño grave en órganos diana en pacientes mexicanos con esclerosis sistémica con afección cutánea difusa. Reumatol Clin 2008; 4(1): 3-7.
6. Navarro C. Afección pulmonary en la esclerosis sistémica. Alveolitis, fibrosis e hipertensión arterial pulmonar. Reumatol Clin 2006; 2 (Supl 3): S16-9.
7. Scully RE, Mark EJ, MacNeely WF, McNeely BU. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 20-1989. A 33-year-old woman with exertional dyspnea and Raynaud’s phenomenon. N Engl J Med 1989; 320(20): 1333-40.
8. Bolster MB, Silver RM. Lung disease in systemic sclerosis (scleroderma). Baillieres Clin Rheumatol 1993; 7(1):79-97.
9. Lamblin C, Bergoin C, Saelens T, Wallaert B. Interstitial lung diseases in collagen vascular diseases. Eur Respir J Suppl. 2001 Sep; 32: 69s-80s.
10. Desai SR, Veeraraghavan S, Hansell DM, Nikolako-polou A, Goh NS, Nicholson AG, et al. CT features of lung disease in patients with systemic sclerosis: comparison with idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia. Radiology 2004; 232(2): 560-7.
11. American Thoracic Society. ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med 2002; 166: 111-7.
12. Ysebrant de Lendonck L, Ocmant A, Roufosse F, Cogan E. Predictive markers for development of severe organ involvement in patients with systemic sclerosis. Ann N Y Acad Sci 2005; 1051: 455-64.
13. Helfrich DJ, Banner B, Steen VD, Medsger TA Jr. Normotensive renal failure in systemic sclerosis. Arthritis Rheum 1989; 32(9): 1128-34.
14. Follansbee WP, Curtiss EI, Medsger TA Jr, Steen VD, Uretsky BF, Owens GR, et al. Physiologic abnormalities of cardiac function in progressive systemic sclerosis with diffuse scleroderma. N Engl J Med 1984; 310(3): 142-8.
15. Valentini G, Vitale DF, Giunta A, Maione S, Gerundo G, Arnese M, et al. Diastolic abnormalities in systemic sclerosis: evidence for associated defective cardiac functional reserve. Ann Rheum Dis 1996; 55(7): 455-60.
16. Ramírez A, Varga J. Pulmonary arterial hypertension in systemic sclerosis: clinical manifestations, pathophysiology, evaluation, and management. Treat Respir Med 2004; 3(6): 339-52.
17. Badui E, Robles E, Hernández C, García Rubí D, Mintz G. Manifestaciones cardiovasculares en la esclerosis sistémica progresiva. Arch Inst Cardiol Mex 1985; 55(3): 263-8.
18. Follansbee WP. The cardiovascular manifestations of systemic sclerosis (scleroderma). Curr Probl Cardiol 1986; 11(5): 241-98.
19. Follansbee WP, Curtiss EI, Medsger TA Jr, Owens GR, Steen VD, Rodnan GP. Myocardial function and perfusion in the CREST syndrome variant of progressive systemic sclerosis. Exercise radionuclide evaluation and comparison with diffuse scleroderma. Am J Med 1984; 77(3): 489-96.
20. Vera O, Jara L. Alteraciones Endócrinas en la Esclerosis Sistémica. Reumatol Clin 2006; 2 Supl 3: S37-41.
21. Rose S, Young MA, Reynolds JC. Gastrointestinal manifestations of scleroderma. Gastroenterol Clin North Am 1998; 27(3): 563-94.
22. Weinstein WM, Kadell BM. The gastrointestinal tract in systemic sclerosis. En: Clements PJ, Furst DE, eds. Systemic sclerosis. 2 ed. Philadelphia: Lippincott Williams & Wilkins; 2004: 293-317.
23. García de la Peña P. Aspectos Clínicos Novedosos en la Esclerodermia. Reumatol Clin 2008; 4 Supl 1: S45-9.
24. Cutolo M, Pizzorni C, Tuccio M, Burroni A, Craviotto C, Basso M, et al. Nailfold videocapillaroscopic patterns and serum autoantibodies in systemic sclerosis. Rheumatology (Oxford) 2004; 43(6): 719-26.
25. Steen VD. Autoantibodies in systemic sclerosis. Semin Arthritis Rheum 2005; 35(1): 35-42.
26. Subcommitte for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Commite. Preliminary criteria for the classification of systemic sclerosis (scleroderma). Arthritis Rheum 1980; 23(5): 581-90.
27. Simeón CP, Armadans L, Fonollosa V, Solans R, Selva A, Villar M, et al. Mortality and prognostic factors in Spanish patients with systemic sclerosis. Rheumatology (Oxford) 2003; 42(1): 71-5.
28. Quadrelli S, Ciallella L, Catalan Pellet A, Molinari L, Salvado A, Auad C, et al. Compromiso pulmonar en esclerosis sistémica. MEDICINA (Buenos Aires) 2007; 67(5): 429-35.
29. Jacobsen S, Halberg P, Ullman S. Mortality and causes of death of 344 Danish patients with systemic sclerosis (scleroderma). Br J Rheumatol 1998; 37(7): 750-5.
30. Ioannidis JP, Vlachoyiannopoulos PG, Haidich AB, Medsger TA Jr, Lucas M, Michet CJ, et al. Mortality in systemic sclerosis: an international meta-analysis of individual patient data. Am J Med 2005; 118(1): 2-10.
31. Chung L, Krishnan E, Chakravarty EF. Hospitalizations and mortality in systemic sclerosis: results from the Nationwide Inpatient Sample. Rheumatology (Oxford) 2007; 46(12): 1808-13.
32. Hesselstrand R, Scheja A, Shen GQ, Wiik A, Akesson A. The association of antinuclear antibodies with organ involvement and survival in systemic sclerosis. Rheumatology (Oxford) 2003; 42(4): 534-40.
33. Vanthuyne M, Blockmans D, Westhovens R, Roufosse F, Cogan E, Coche E, et al. A pilot study of mycophenolate mofetil combined to intravenous methylprednisolone pulses and oral low-dose glucocorticoids in severe early systemic sclerosis. Clin Exp Rheumatol 2007; 25(2): 287-92.
34. Denton CP, Black CM. Targeted therapy comes of age in scleroderma. Trends Immunol 2005; 26(11): 596-602.
35. Antoniou KM, Wells AU. Scleroderma lung disease: evolving understanding in light of newer studies. Curr Opin Rheumatol 2008; 20(6): 686-91.

