










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkRevista Médica del Uruguay
versión On-line ISSN 1688-0390
Rev. Méd. Urug. vol.22 no.3 Montevideo set. 2006
Fiebre mediterránea familiar: una afección frecuentemente subdiagnosticada
Dres. Fernando Mañé Garzón*, Víctor Raggio†
Departamento de Genética, Facultad de Medicina. Universidad de la República
Resumen
La fiebre mediterránea familiar (FMF) es una enfermedad inflamatoria crónica, hereditaria, de herencia autosómica recesiva, causada por mutaciones en el gene denominado MEFV. Se caracteriza por episodios recurrentes de fiebre e inflamación multisistémica. Se trata de una enfermedad frecuente en descendientes de poblaciones mediterráneas, del norte de África, Israel, Turquía, Armenia y países árabes. Se presenta el caso de un paciente, descendiente de armenios, con semiología y evolución características. Se analiza el diagnóstico, pronóstico, tratamiento y el asesoramiento genético correspondiente.
Palabras clave: Fiebre MediterrÁnea Familiar - diagnóstico.
Fiebre MediterrÁnea Familiar - terapia.
Fiebre MediterrÁnea Familiar - genética.
Fiebre MediterrÁnea Familiar - etiología.
* Ex Profesor de Clínica Pediátrica, Profesor Emérito de la Facultad de Medicina.
† Profesor Adjunto del Departamento de Genética, Facultad de Medicina.
Correspondencia: Dr. Víctor Raggio
Departamento de Genética, Facultad de Medicina
Gral. Flores 2125. CP 11800. Montevideo, Uruguay.
E-mail: vraggio@fmed.edu.uy
Recibido: 29/8/05.
Aceptado: 8/6/06.
Introducción
La fiebre mediterránea familiar (FMF) es una enfermedad inflamatoria crónica, hereditaria, de herencia autosómica recesiva que se caracteriza por episodios recurrentes de dolor abdominal y varias formas de serositis acompañados de fiebre elevada. Ha recibido varios nombres en la literatura anglosajona y francesa, siendo los más usados: "Benign paroxysmal peritonitis", "Recurrent Polyserositis", "Periodic Disease" y "Maladie periodique".
La enfermedad afecta predominantemente a poblaciones procedentes de la región mediterránea: judíos norafricanos (judíos no asquenazis en general), armenios, turcos y árabes. La frecuencia de portadores se ha calculado en 1:5 a 1:7 en algunas de estas poblaciones, llegando la frecuencia de enfermos (homocigotos) a 1/200 individuos. Algunos autores señalan, a partir de su experiencia clínica en poblaciones de Estados Unidos, que los descendientes de italianos también tienen un riesgo aumentado para esta afección(1 ).
El inicio de los síntomas es, generalmente, en las primeras dos décadas de la vida y es excepcional que comiencen luego de los 30 años. La enfermedad evoluciona característicamente en empujes y remisiones; los ataques, como en todas las enfermedades cíclicas, pueden estar separados por días o años, y pueden ser desencadenados por sucesos ambientales, como, por ejemplo, el estrés y la actividad física extrema. Los hechos más frecuentes de estas crisis inflamatorias son los siguientes: 1) fiebre recurrente, presente en todas las crisis y que puede llegar a 40ºC; 2) episodios de dolor abdominal agudo, el cual muchas veces es difícil de diferenciar con síndromes quirúrgicos, por lo que se realizan frecuentemente laparotomías "en blanco"; 3) otras manifestaciones de serositis: pleuritis y pericarditis; 4) artritis, en la mayoría de los casos se trata de una oligoartritis aguda, asimétrica, que se resuelve espontáneamente. Otras manifestaciones más raras incluyen: mialgia febril, eritema tipo erisipela, vasculitides: púrpura de Henoch-Schönlein y poliarteritis nodosa, meningitis recidivante de Mollaret, esplenomegalia, orquitis, etcétera.
La incidencia de esta afección en nuestra población, que cuenta con un importante contingente inmigratorio de origen mediterráneo, no debe ser desestimada, por lo que consideramos interesante presentar este caso demostrativo.
Caso clínico
Escolar, sexo masculino, de 9 años de edad. Sus cuatro abuelos son de procedencia armenia.
Es producto de segunda gestación, con embarazo bien controlado, mal tolerado por amenaza de aborto, pero que evoluciona satisfactoriamente con reposo. Parto vaginal a término, recién nacido de peso adecuado para la edad gestacional, buen crecimiento y desarrollo. Hernia inguinal bilateral, operada a los 2 años de edad. Actualmente cursando cuarto año de primaria. Comienza a los 6 meses de vida con episodios febriles, a predominio nocturno, de inicio brusco, alcanzando picos de hasta 40ºC axilar, acompañados de chuchos; estos episodios duraban aproximadamente 24 horas pero seguidos, cada uno de ellos, por un período de entre 15 y 20 días con picos febriles. El tiempo de intercrisis duraba aproximadamente tres meses, totalizando ocho episodios en los primeros dos años de vida. Los ataques se caracterizan, además, por las alteraciones serológicas que se detallan: leucocitosis de entre 20-24 x 103 sin desviación a izquierda, velocidad de eritrosedimentación (VES) de entre 120-140 mm/h, proteína C reactiva positiva, aumento leve de globulinas a2 e hiperplaquetosis. Asimismo, se evidenció anemia ferropénica con reticulocitosis baja y depósitos de hierro conservados.
Estudiado intensamente a fin de determinar el origen de estos episodios: múltiples hemocultivos, urocultivos, exudados faríngeos y consultas con otorrinolaringólogo: todos negativos; reacción de Paul Bunell, anticuerpos antivirus de Ebstein Barr (EBV), toxoplasma, citomegalovirus, clamidias, leptospira y rickettsias: también negativos. Mielocultivo, que fue negativo, y mielograma que mostró: celularidad aumentada con las tres series presentes, con hiperplasia de la series mielogranulocítica, y megacariocítica, sin alteraciones en la serie eritroide o linfoplasmocitaria, no infiltración por células inmaduras o extrahematopoyéticas; células LE, anticuerpos anti ADN y factor reumatoideo, negativos. Múltiples radiografías de tórax y de senos faciales, normales. Ecocardigrama que no mostró masas intracavitarias, vegetaciones valvulares, ni derrame pericárdico. Ecografía abdominal, tomografía computarizada de cráneo, cuello, tórax, abdomen y pelvis, normales.
En la evolución persiste con este tipo de ataques, aproximadamente con la misma frecuencia, y teniendo como máximo períodos de seis meses asintomático. A medida que el niño crece, los padres notan otros elementos acompañantes de los ataques como: artralgias, fundamentalmente a nivel de rodillas, sin otros elementos de inflamación, erupciones máculo-eritematosas a nivel de tronco, y dolor abdominal intenso, difuso, no cólico, que no se acompaña de vómitos. No se relatan elementos sugestivos de afectación de otras serosas. Los padres notan, como desencadenantes de los episodios, cuadros respiratorios altos o eventos estresantes para el niño. Evaluación actual de la función renal: normal.
Comentarios
Se sospecha el diagnóstico de FMF ante la presencia de episodios febriles recurrentes, con fiebre de hasta 40ºC, acompañados de peritonitis, sinovitis, eritema y elementos serológicos de reacción de fase aguda, sin causa infecciosa, autoinmune o neoplásica demostrada, que aparecen en un paciente perteneciente a un grupo étnico de riesgo, en este caso descendiente de armenios. En su evolución no ha aparecido la principal y más grave manifestación de la afección: la amiloidosis renal de tipo AA. La misma ocurre luego de varios años, especialmente en pacientes no tratados.
La FMF está producida por mutaciones en el gen MEFV, situado en el Locus 16p13, que consta de diez exones. Los alelos mutantes codifican para una proteína menos activa debido a mutaciones puntuales, principalmente de cambio de sentido. Se han identificado cerca de 90 mutaciones(2), siendo las más frecuentes: M694V, V726A (exón 10) y E148Q (exón 2). La mutación M694V muestra una asociación significativa, tanto en homo como en heterocigosis, con el desarrollo de amiloidosis renal. La mutación compuesta V726A-E148Q también implica una evolución más severa con mayor tendencia a la amiloidosis renal(3).
Para explicar la alta frecuencia de estos alelos mutantes en determinadas poblaciones se invocan tanto fenómenos de efecto fundador, deriva génica, como posibles ventajas selectivas de determinadas combinaciones de heterocigotos(4).
El gen normal codifica un transcripto de 3.7 Kb que es expresado exclusivamente en los granulocitos maduros y los monocitos. La proteína codificada por este gen ha sido llamada Pirina por el International FMF Consortium y Marenostrin por el French FMF Consortium. Contiene 781 aminoácidos, y su función normal es probablemente asistir en la regulación de la respuesta inflamatoria al desactivar la respuesta inmune. Se ha encontrado un aumento de la expresión de los genes de varias citoquinas en los pacientes con FMF, lo que podría considerarse un estado de preactivación de células inflamatorias, por lo que se propone la persistencia de un estado de inflamación subclínica durante la remisión(5). Sin embargo, se desconoce el mecanismo exacto por el que las mutaciones en MEFV resultan en una alteración de las repuestas inflamatorias. Se ha propuesto que regula la respuesta inflamatoria a nivel del citoesqueleto leucocítico, lo que explicaría el efecto terapéutico de la colchicina(6). Posteriormente se encontró una variante de splicing de la proteína, denominada MEFV-d2 que se localiza en el núcleo, por lo que podría tratarse de un factor nuclear que actúa en la regulación de la expresión génica(7).
El diagnóstico se puede confirmar a nivel molecular, ya sea en forma indirecta por análisis de ligamiento a microsatélites marcadores, o en forma directa detectando las mutaciones más frecuentes según el origen étnico del paciente(8), o por secuenciación directa de los exones(9 ). El conocimiento de las mutaciones causales tiene, además, importancia pronóstica y como guía del tratamiento ya que los portadores de determinadas mutaciones son los que tienen mayor riesgo de sufrir amiloidosis renal.
Se trata seguramente de una enfermedad subdiagnosticada, ya que si no se tiene en cuenta la genética de poblaciones que explica la aparición de la misma y no existen antecedentes familiares sugestivos, es difícil que se sospeche(10 ). Se estima que en Uruguay viven unos 15.000 descendientes directos de armenios, por lo que si asumimos una frecuencia de heterocigotos de uno en 14 individuos descendientes de armenios(1), debería haber cerca de 100 casos sólo en esta comunidad.
En este caso no aparecen antecedentes que hagan sospechar la enfermedad en la familia, lo que es frecuente en enfermedades recesivas, pero no debe olvidarse interrogar al respecto, ya que se pueden obtener datos diagnósticos muy relevantes.
El primer caso publicado en Uruguay fue hecho por Canzani, Fisher y Álvarez Martínez hace 40 años(11). Destacan los autores el carácter predominante étnico y familiar de la afección y describen la frecuencia alta en algunas poblaciones; lamentablemente, los autores no analizan el origen poblacional de su paciente para sustentar el diagnóstico. Sí destacan la ausencia de antecedentes familiares, como frecuentemente se observa en enfermedades recesivas. En la lectura del trabajo se aprecian dificultades en la definición de la entidad al desconocerse la causa. Se debe tener en cuenta que la actual clasificación de los síndromes inflamatorios hereditarios es muy reciente (fines de la década de 1990).
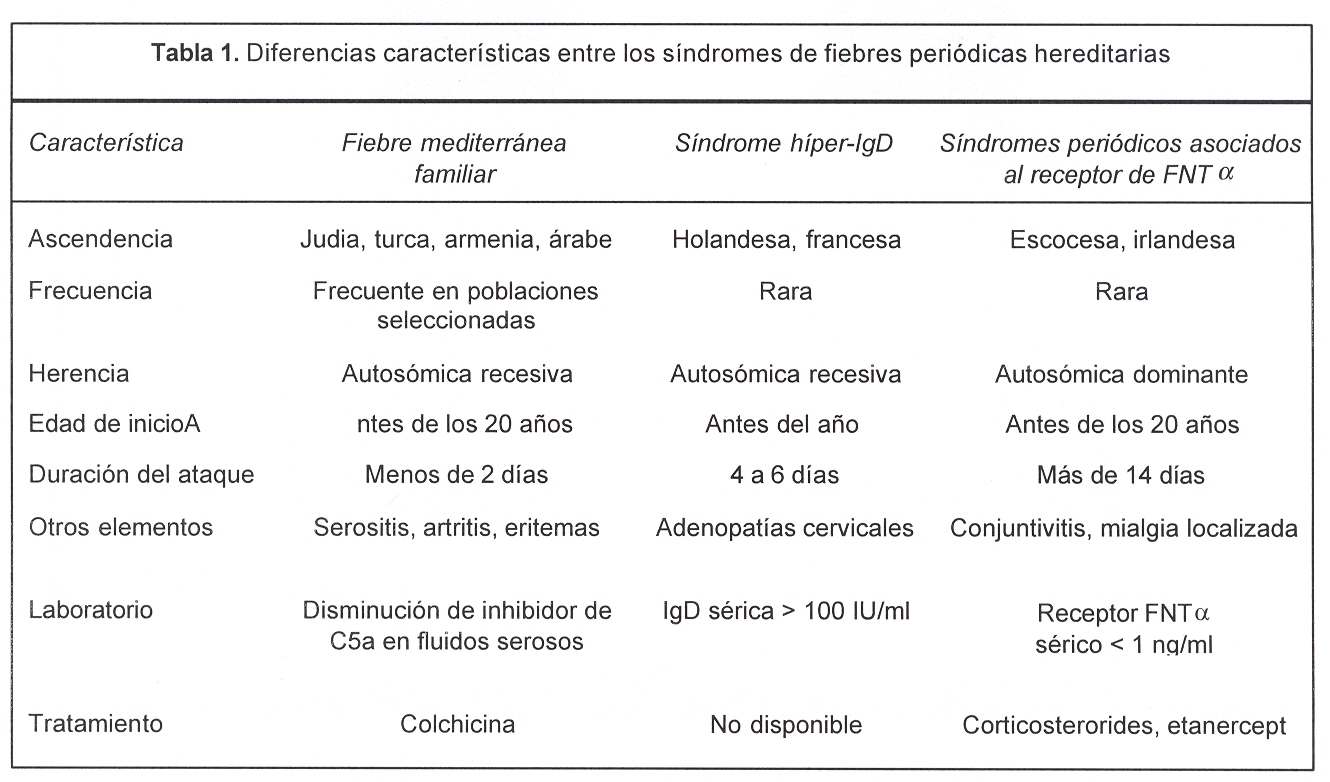
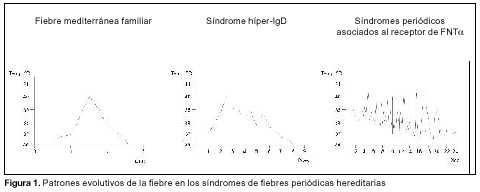
En relación con el diagnóstico diferencial se deben plantear otras fiebres periódicas familiares, actualmente denominadas "enfermedades autoinflamatorias"(12,13) (figura 1 y tabla 1)(14,15). Ellas son: síndrome de hiperinmunoglobulinemia D: enfermedad autosómica recesiva, rara, causada por mutaciones de pérdida de función en el gen de la mevalonato quinasa, enzima de la vía biosintética del colesterol. Se desconoce cómo un defecto del metabolismo del colesterol produce una enfermedad inflamatoria recurrente. Hay algunos indicios de que respondería favorablemente al tratamiento con simvastatina(16); los síndromes periódicos asociados al receptor del factor de necrosis tumoral a tipo 1 (FNTa), anteriormente denominados como Familial Hibernian Fever o simplemente fiebre periódica familiar: son de herencia autosómica dominante y están causados por mutaciones en el gene TNFR1 (12p13.2) que codifica el receptor del FNTa. Responden a altas dosis de prednisona y la terapéutica específica es etanercept, proteína recombinante que consiste en la fusión de dos copias de la porción extracelular de unión al ligando del receptor con la porción Fc de IgG, esta proteína se une al FNTa circulante y atenúa sus efectos biológicos; la urticaria familiar desencadenada por el frío (UF): heredada de forma autosómica dominante, la exposición al frío desencadena un rash maculopapular, no pruriginoso, a veces asociado con fiebre, artralgias, mialgias, conjuntivitis y cefaleas. El síndrome de Muckle-Wells (SMW): de herencia autosómica dominante, similar al descripto anteriormente, pero los síntomas no son desencadenados por el frío y desarrollan frecuentemente sordera neurogénica progresiva. Tanto el SMW como la UF han sido mapeados recientemente en 1q44(17), y se ha demostrado que se deben a mutaciones en el gene CLAS1, el cual codifica una proteína que por sus características estructurales tendría un rol inflamatorio(18).
Por supuesto que en algunos casos, especialmente los que se presentan con dolor abdominal, se deben descartar síndromes agudos de abdomen o enfermedades ginecológicas. En aquellos pacientes con dolor articular, el diagnóstico diferencial incluye artritis reumatoidea, fiebre reumática, artritis séptica y enfermedades del colágeno.
Si bien en algunos casos son más prolongados, en nuestro paciente los ataques agudos revierten espontáneamente y sin secuelas luego de 24-48 horas. Desde hace dos años los padres tratan los ataques con "pulsos" de corticoides, reduciendo la dosis paulatinamente, con lo cual se logra una remisión de todos los síntomas. No se han obtenido respuestas favorables con antiinflamatorios no esteroideos. Se inició el tratamiento de rigor con colchicina: 0,5 mg / día, desde los 2 años, administrada de forma irregular por intolerancia digestiva; por este mismo motivo no se ha podido aumentar la dosis a la indicada de 1-2 mg/d. El tratamiento con colchicina se debe intentar siempre dado que, en general, previene los ataques agudos, y aun sin lograr este efecto, disminuye el riesgo de amiloidosis. El tratamiento se debe continuar de por vida. A las dosis que se utiliza, la colchicina no afecta el crecimiento ni el desarrollo e incluso permite retomar el crecimiento ponderal en aquellos niños afectados de ataques severos o frecuentes al disminuir la frecuencia de los mismos(19).
Al tratarse de una enfermedad que tiene un modo de herencia autosómico recesivo, ambos padres del propósito son considerados portadores (heterocigotos) obligados. Sin embargo, en poblaciones con alta frecuencia de portadores y en particular donde exista una alta tasa de matrimonios consanguíneos, es posible que niños enfermos nazcan de uniones entre un individuo afectado (homocigoto) con un portador, o incluso entre dos individuos afectados. Si ambos padres son heterocigotos, el riesgo de recurrencia para los hermanos es de 25%. El diagnóstico prenatal es posible en embarazos de parejas con mutaciones conocidas en el gen MEFV.
Summary
Familial Mediterranean Fever (FMF) is an inherited autosomic recessive and inflammatory chronic disease caused by MEFV gene mutation. Symptoms are comprised over recurrent episodes of fever and multisystemic inflammation. Prevalence is higher in Mediterranean population, North Africa, Israel, Turky, Armenia and Arab countries. A case report of an Armenian descent man with characteristic signs and development. Diagnosis, prognosis, treatment and subsequent genetic advise are analyzed.
Résumé
La fièvre méditerranéenne familiale (FMF) est une maladie inflammatoire chronique, héréditaire, à transmission autosomique récessive, causée par des mutations au gène MEFV. Elle s’exprime par des crises fébriles brèves et inflammation multi systémique. Il s’agit d’une maladie fréquente chez des sujets originaires du pourtour méditerranéen, du nord de l’Afrique, l’Israël, la Turquie, l’Arménie et des pays arabes. On présente le cas d’un patient descendant d’Arméniens, avec une sémiologie et une évolution caractéristiques. On analyse le diagnostic, le pronostic, le traitement et les données génétiques.
Resumo
A febre mediterrânea familiar (FMF) é uma doença inflamatória crônica, hereditária, de herença autossômica recessiva, causada por mutações no gen MEFV. Caracteriza-se por episódios recorrentes de febre e inflamação multissistêmica. Trata-se de uma patologia frequente em descendentes de populações mediterrâneas do norte da África, Israel, Turquia, Armênia e países árabes. Descreve-se o caso de um paciente, descendente de armênios, com semiologia e evolução características. Faz-se a análise do diagnóstico, prognóstico, tratamento e do correspondente assessoramento genético.
Bibliografía
1. Kastner D. Familial Mediterranean fever: the genetics of inflammation. Hosp Pract (Minneap) 1998; 33 (4): 131-4, 139-40, 143-6.
2. Touitou I, Lesage S, McDermott M, Cuisset L, Hoffman H, Dode C, et al. Infevers: an evolving mutation database for auto-inflammatory syndromes. Hum Mutat 2004; 24(3): 194-8.
3. Gershoni-Baruch R, Brik R, Shinawi M, Livneh A. The differential contribution of MEFV mutant alleles to the clinical profile of familial Mediterranean fever. Eur J Hum Genet 2002; 10(2): 145-9.
4. Schaner P, Richards N, Wadhwa A, Aksentijevich I, Kastner D, Tucker P, et al. Episodic evolution of pyrin in primates: human mutations recapitulate ancestral amino acid states. Nat Genet 2001; 27(3): 318-21.
5. Notarnicola C, Didelot MN, Seguret F, Demaille J, Touitou I. Enhanced cytokine mRNA levels in attack-free patients with familial Mediterranean fever. Genes Immun 2002; 3(1): 43-5.
6. Mansfield E, Chae JJ, Komarow HD, Brotz TM, Frucht DM, Aksentijevich I, et al. The familial Mediterranean fever protein, pyrin, associates with microtubules and colocalizes with actin filaments. Blood 2001; 98(3): 851-9.
7. Papin S, Duquesnoy P, Cazeneuve C, Pantel J, Coppey-Moisan M, Dargemont C, et al. Alternative splicing at the MEFV locus involved in familial Mediterranean fever regulates translocation of the marenostrin/pyrin protein to the nucleus. Hum Mol Genet 2000; 9(20): 3001-9.
8. Eisenberg S, Aksentijevich I, Deng Z, Kastner DL, Matzner Y. Diagnosis of familial Mediterranean fever by a molecular genetics method. Ann Intern Med 1998; 129(7): 539-42.
9. Grateau G, Pecheux C, Cazeneuve C, Cattan D, Dervichian M, Goossens M. Clinical versus genetic diagnosis of familial Mediterranean fever. Q J M 2000; 93 (4): 223-9.
10. Stewart L, Tolmie J, Galea P, Touitou I. Familial Mediterranean Fever in a cold climate: read The Lancet. Lancet 2000; 356 (9248): 2154.
11. Canzani R, Fischer TM, Álvarez-Martínez J. Fiebre Mediterránea Familiar. Día Méd Urug 1965; XXXII (388): 597-605.
12. Touitou I, Notarnicola C, Grandemange S. Identifying mutations in autoinflammatory diseases: towards novel genetic tests and therapies?. Am J Pharmacogenomics 2004; 4(2): 109-18.
13. Horcada Rubio ML, Delgado Beltran C, Armas Ramírez C. Autoinflammatory disorders: a new concept in hereditary periodic fever syndromes. An Med Interna 2004; 21(3): 143-7.
14. Drenth JP, van-der-Meer JW. Hereditary periodic fever. N Engl J Med 2001; 345 (24): 1748-57.
15. Drenth JP, van Der Meer JW. Periodic fevers enter the era of molecular diagnosis. BMJ 2000; 320: 1091-2.
16. Simon A, Drewe E, van-der-Meer JW, Powell RJ, Kelley RI, Stalenhoef AF, et al. Simvastatin treatment for inflammatory attacks of the hyperimmunoglobulinemia D and periodic fever syndrome. Clin Pharmacol Ther 2004; 75(5): 476-83.
17. Cuisset L, Drenth JP, Berthelot JM, Meyrier A, Vaudour G, Watts RA, et al. Genetic linkage of the Muckle-Wells syndrome to chromosome 1q44. Am J Hum Genet 1999; 65(4): 1054-9.
18. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 2001; 29(3): 301-5.
19. Shohat M. Familial Mediterranean Fever. In: GeneReviews. Medical Genetics Information Resource (database online). Seattle: University of Washington, 2004. Obtenideo de: http://www.genetests.org/query?dz=fmf (visto 20/7/2005).

