










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkRevista Médica del Uruguay
versión On-line ISSN 1688-0390
Rev. Méd. Urug. vol.21 no.2 Montevideo jun. 2005
Malformación adenomatosa quística pulmonar.
Diagnóstico prenatal y evolución de un caso
Dres. Marcelo De Agostini Cicognani*, Ricardo Levy Mizraji†
Hospital Evangélico de Montevideo. Departamento de Imagenología.
Sección Ecografía Ginecotocológica
Resumen
Se presenta el caso clínico de una mujer que cursa su segundo embarazo, donde se le diagnostica ecográficamente al feto una malformación adenomatosa quística pulmonar (MAQP), a las 19 semanas.
La evolución del embarazo fue sin complicaciones, llegando a término y sin requerir tratamientos intrauterinos ni estudios especiales.
El recién nacido fue dado de alta con buena evolución y asintomático.
Dado lo infrecuente de la enfermedad, así como su evolución y los pocos casos de diagnóstico prenatal reportados, se decide su presentación.
Palabras clave: MALFORMACIÓN ADENOMATOIDE QUÍSTICA CONGÉNITA DEL
PULMÓN - diagnóstico.
MALFORMACIÓN ADENOMATOIDE QUÍSTICA CONGÉNITA DEL
PULMÓN - ultrasonografía.
ULTRASONOGRAFÍA PRENATAL.
* Médico ginecotocólogo y ecografista ginecoobstétrico, ex Residente y Asistente de Clínica Ginecotocológica. Asistente Grado 2 de la Unidad de Ecografía de la Clínica Ginecotocológica "B", Hospital de Clínicas, Facultad de Medicina. Universidad de la República. Montevideo. Uruguay
† Médico Ginecotocólogo y ecografista ginecoobstétrico.
Correspondencia: Dr. Marcelo De Agostini
26 de Marzo 1180, CP 11300, Montevideo, Uruguay
E-mail: drmda@hotmail.com
Recibido: 2/2/04.
Aceptado: 28/12/04.
Introducción
Hasta la fecha hay reportados en la literatura internacional menos de 200 casos de malformación adenomatosa quística pulmonar (MAQP)(1-3), de ahí lo interesante del hallazgo, así como las escasas y poco eficaces, hasta el momento, recursos terapéuticos disponibles.
Es una enfermedad excepcional descripta por primera vez por Ch’in y Tang en 1949. Comprende 25% de las lesiones pulmonares congénitas halladas en autopsias(4).
El primer reporte de diagnóstico prenatal de esta enfermedad fue realizado en 1975 por Garret y colaboradores(5). La mayor casuística a la fecha la describe N. Scott Adzick, con más de 175 casos de diagnóstico prenatal(24).
Se transmite con un patrón esporádico, sin que se conozca asociación a teratogénicos(1).
Es más frecuente en el varón que en la mujer, en razón de 7 a 1(6).
Son tumores compresivos, responsables de hipoplasia pulmonar o de anasarca, o ambas, en relación con un problema en la circulación de retorno.
Se trata de tumores benignos quísticos del pulmón, que suelen localizarse en un lóbulo (habitualmente es unilateral). En 95% de los casos se limita a un lóbulo o segmento.
Existen varios tipos: macroscópico, histológico y ecográfico, ordenados en tres categorías según la clasificación de Stocker.
Clasificación de Stocker y colaboradores (1977)(26)
Tipo I: quistes de gran tamaño (2 a 10 cm).
Tipo II: quistes de menor tamaño (0,5 a 2 cm).
Tipo III: microquistes (menores de 0,5 cm).
Existe otra clasificación –también utilizada– que es la de Adzick y colaboradores(7).
Clasificación de Adzick y colaboradores
MAQP macroquística (quistes mayores o iguales a 5 mm).
MAQP microquística (quistes menores de 5 mm).
Esta clasificación se utiliza para el diagnóstico prenatal(27). Histológicamente se trata de una lesión de tipo hamartomatosa(8), caracterizada por un crecimiento excesivo de los bronquiolos terminales y una marcada ausencia de alvéolos, que forman quistes de diverso tamaño y alvéolos anormales.
Las formas quísticas aisladas tienen mejor pronóstico y su evolución es más lenta (es el caso que reportamos).
Puede regresar espontáneamente hasta en casi 10% de los casos.
Caso clínico
Se trata de una paciente de 29 años, secundigesta, nulípara (con el antecedente de un aborto espontáneo), que cursando la semana 19 de gestación se le diagnostica, durante la realización del estudio ecográfico morfológico fetal, a nivel del pulmón derecho del feto, la presencia de por lo menos cuatro imágenes quísticas de entre 12 mm y 19 mm de diámetro (macroquistes), agrupadas, de contenido líquido denso y homogéneo (hipoecogénicas), con halo ecogénico limitante, y que desplazaban el mediastino.
El resto del parénquima pulmonar, así como el pulmón contralateral, no presentaba alteraciones de su ecogenicidad y era de aspecto ecográfico normal. No se destacaban otras alteraciones anatómicas en el resto de la exploración y la antropometría fetal era la adecuada.
Estos hallazgos hicieron pensar en la enfermedad que estamos tratando luego de descartar los diagnósticos diferenciales ya analizados.
La evolución de dichas lesiones fue sin importantes modificaciones de volumen durante toda la gestación, persistiendo similar el tamaño y proporcionalmente, con buen desarrollo del parénquima pulmonar restante.
Se trataba de una MAQP tipo I de acuerdo con la clasificación de Stocker y colaboradores.
Fue controlada con equipo interdisciplinario (obstetra, ecografista, cirujano pediátrico, genetista y psicólogo), se realizó ecocardiograma fetal, que fue normal, destacado únicamente el desplazamiento mediastinal determinado por los quistes; no se le aconsejó la realización de cariotipo fetal, tras la interconsulta con genetista, ya que la pareja no deseaba arriesgar la vida de su futuro hijo tras una punción, y el embarazo transcurrió sin otos incidentes y con un buen desarrollo hasta el término, en que se le realizó una operación cesárea por indicación materna (estado hipertensivo del embarazo).
El recién nacido con test de Apgar 9/10, tuvo un peso de 3.500 g y una buena evolución neonatal, sin requerir tratamientos especiales.
Fue dado de alta con indicación de realizar resonancia nuclear magnética a los 30 días de vida, para determinar si tiene enfermedad residual pulmonar.
Aspectos ecográficos
Ecográficamente se presenta como una masa de quistes grandes (I) o pequeños (II), o bien muestra un aspecto sólido-ecogénico generado por el patrón microquístico (III).
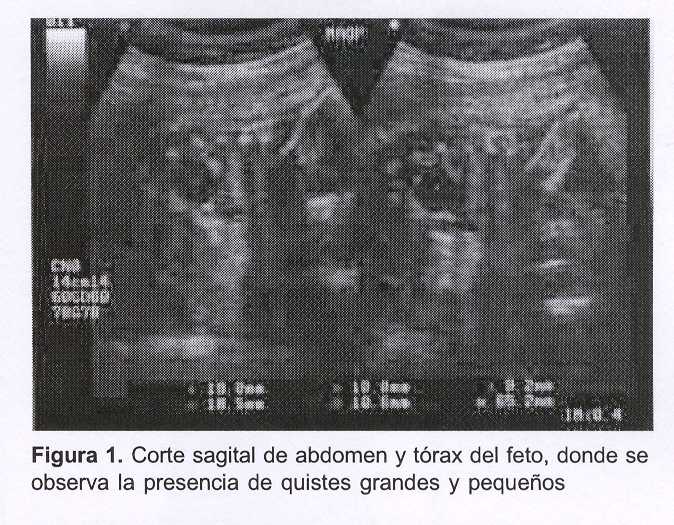
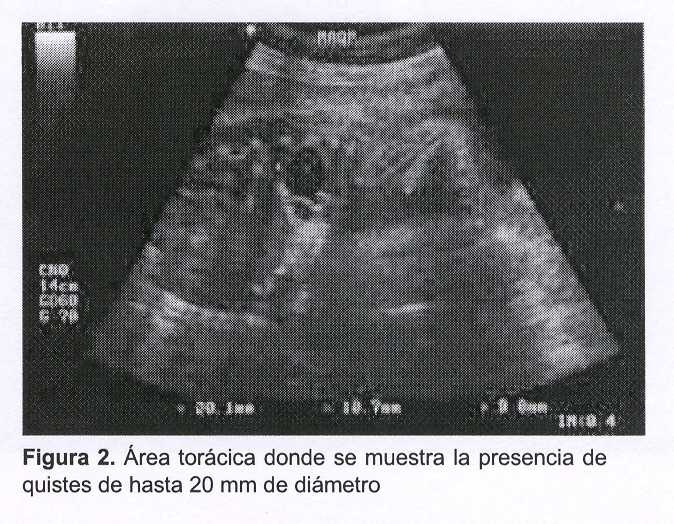
El ultrasonido de alta resolución es fundamental para aclarar el diagnóstico (figuras 1 y 2).
Como diagnósticos diferenciales a los quistes broncogénicos hay que plantearse: el secuestro pulmonar, que es una masa de tejido pulmonar anormal que no se comunica con el árbol bronquial y posee vascularización propia anormal, en estos casos el eco doppler color es de gran utilidad para el diagnóstico(9); los quistes de duplicación esofágica; los quistes mesentéricos, y el teratoma quístico.
Discusión y conclusiones
Malformaciones asociadas
Si bien la MAQP es una condición habitualmente aislada, se han hallado asociaciones en 20% de los casos. Las más frecuentes son uropatías (agenesia o disgenesia renal), malformaciones anorrectales, hernia diafragmática y anomalías cardíacas(28).
También fueron descriptas alteraciones del sistema nervioso central, onfalocele y gastrosquisis(10,11).
Se ha observado, a su vez, un leve aumento en la incidencia de cromosomopatías en los fetos portadores de una MAQP(12). No hallamos asociaciones en el caso reportado.
Pronóstico
La MAQP desde el punto de vista pronóstico y ya en el segundo trimestre de la gestación, puede regresar espontáneamente en forma total en 6% de los casos(13-15), puede evolucionar al polihidramnios y a la aparición de hidrops fetal, o pueden persistir los quistes hasta el nacimiento, momento en que pueden ser extirpados(16).
Son factores de buen o mal pronóstico prenatal el tamaño de la lesión, el grado de rechazo del mediastino y el desarrollo de hidrops o hidramnios, o ambos(6,13,17).
En cuanto al pronóstico al nacer, la radiología, la tomografía axial computarizada y la resonancia nuclear magnética determinaran si hay enfermedad residual pulmonar. Puede ser asintomático o determinar infecciones respiratorias recurrentes(17).
Puede desarrollar lesiones malignas, es por eso que se aconseja la resección de los macroquistes al nacer, aunque sean asintomáticos(18,19).
La malformación adenomatosa quística pulmonar unilateral y que no desarrolla hidrops fetal, tal como fue el caso que estamos presentando, tiene un pronóstico favorable y se aconseja mantener, en principio, una conducta expectante(20).
Conducta prenatal y terapia in útero
Por supuesto que hay que confirmar el hallazgo ecográfico en un centro de referencia, realizar detalladamente una ecografía estructural fetal que incluya ecocardiograma para descartar asociaciones. Eventualmente puede realizarse el estudio del cariotipo fetal mediante amniocentesis, dada la asociación a cromosomopatías de esta enfermedad. A partir de aquí es fundamental el seguimiento ecográfico seriado, documentando la evolución, tamaño de las lesiones, grado de desplazamiento mediastinal, hidrops, hidramnios, etcétera.
La interconsulta con cirujano pediátrico es necesaria por si es preciso un tratamiento quirúrgico in útero o al nacer.
Hay que planificar el nacimiento en un centro de referencia de tercer nivel.
Estos fueron los pasos que se siguieron en el caso que presentamos.
No fue necesario realizar un tratamiento in útero, ya que no se evolucionó al hidrops fetal.
Los casos que requirieron tratamiento in útero y fueron reportados se realizaron con feto hidrópico y en los que no se había alcanzado aún la maduración pulmonar(21).
Esto fue descrito en el Centro de Tratamiento Fetal de la Universidad de California, San Francisco.
Las opciones terapéuticas son la aspiración de los quistes de gran tamaño(22,23), la derivación tóraco-amniótica de un quiste dominante(24), y la resección quirúrgica de la masa(25).
La cirugía fetal se debe realizar en centros de referencia. Existen pocos en el mundo, uno de ellos en el Centro de Tratamiento Fetal de la Universidad de California, San Francisco, donde su actual jefe, el Prof. Dr. Michael Harrison establece claramente que estos tratamientos deben ser realizados por equipos interdisciplinarios capacitados y entrenados previamente en cirugía experimental y en donde las consideraciones éticas y legales deberán ser tenidas en cuenta.
Se ha planteado también la destrucción hipertérmica de las lesiones con láser Nd:Yag, en el tipo III, hasta el momento experimental, existiendo un solo caso reportado a la fecha. Este tipo presenta un mal pronóstico y la evolución natural es hacia la anasarca, con una alta probabilidad de muerte fetal (38%).
Si se comprueba anasarca fetal antes de la semana 32, también puede plantearse la realización de una lobectomía pulmonar fetal por histerotomía.
Reflexión final
El feto es una persona y puede estar enfermo, intoxicado, contaminado o ser portador de una neoplasia. Nuestro objetivo es prevenir el desarrollo de enfermedades, prevenir la aparición de complicaciones asociadas a la prematurez y, en ocasiones, considerar el tratamiento paliativo o curativo in útero (infecciones, tumores, uropatías, anemia, etcétera). Pero es necesario recordar siempre que debemos evitar la yatrogenia.
La medicina fetal es una nueva especialidad médica (médica y quirúrgica, preventiva y curativa). A la madre se la somete, en oportunidades, en demasía, y muchas veces no está enferma, de ahí la necesidad de discutir mucho la elección del tratamiento más adecuado para el feto. Hay que tener en cuenta siempre los efectos secundarios para la madre, los beneficios del tratamiento intrauterino del feto y los riesgos de un parto prematuro.
Es de esperar que para diagnósticos precoces de casos similares siempre se actúe con cautela y se realice un seguimiento y tratamiento lo más adecuados posible, evitando algunas complicaciones e intentando prevenir otras (hipoplasia pulmonar remanente, insuficiencia cardíaca congestiva, prematurez, etcétera).
Debemos resaltar que fue fundamental el apoyo y el asesoramiento psicológico de la pareja y de su entorno familiar, dirigido por el equipo médico tratante, ya que un diagnóstico prenatal de este tipo plantea innumerables dudas y temores, y puede desencadenar una serie de decisiones muchas veces inapropiadas.
Summary
A 19-week embryon (mother coursing her second pregnancy) was diagnosed by ecography with a cystic adenomatoid pulmonary malformation (CAPM).
No complications were found during pregnancy evolution. Pregnancy was carried to term without need of intrauterine treatments.
The newborn was asymptomatic and discharged to home. The case is presented due its rareness among prenatal diagnosis reports.
Résumé
On présente le cas clinique d’une femme pendant sa deuxième grossesse au cours de laquelle on fait un diagnos-tic au foetus de malformation adénomatose kystique pulmonaire (MAKP) à la 19è semaine.
L’évolution de la grossesse a été normale; pas besoin d’études spéciales ni de traitements intra-utérins.
Le nouveau-né est parti sans symptômes.
Étant donné que cette maladie est très rare et qu’il y en a peu de cas de diagnostic prénatal repportés, on a décidé de le présenter.
Bibliografía
1. Adzick NS, Harrison MR, Glick PL, Golbus MS, Anderson RL, Mahony BS, et al. Fetal cystic adenomatoid malformation: prenatal diagnosis and natural history. J Pediatr Surg 1985; 20(5): 483-8.
2. Bruner JP, Jarnagin BK, Reinisch L. Fetal congenital cystic adenomatoid malformation of the lung. Fetal diagn Ther 2000; 15(6): 359-63.
3. Cha I, Adzick NS, Harrison MR, Finkbeiner WE. Fetal congenital cystic adenomatoid malformation of the lung: a clinic pathologic study of eleven cases. Am J Surg Pathol 1997; 21(5): 537-44.
4. Merkus PJ, ten-Have-Opbroek AA, Quanjer PH. Human lung growth: a review. Pediatr Pulmonol 1996; 21(6): 383-97.
5. Garret WJ, Kossoff G, Lawrence R. Gray scale echography in the diagnosis of hydrops due to fetal lung tumor. J Clin Ultrasound 1975; 3(1): 45-50.
6. Pezzuti RT, Isler RJ. Antenatal ultrasound detection of cystic adenomatoid malformation of lung. Report of a case and review of the recent literature. J Clin Ultrasound 1983; 11(6): 342-6.
7. Adzick NS, Harrison MR, Glick PL, Golbus MS, Anderson RL, Mahony BS, et al. Fetal cystic adenomatoid malformation: prenatal diagnosis and natural history. J Pediatr Surg 1985; 20(5): 483-8.
8. Ch’in KY, Tang MY. Congenital adenomatoid malformation of one lobe of a lung with general anasarca. Arch Pathol 1949; 48: 155-71.
9. Sauerbrei E. Lung sequestration. Duplex Doppler diagnosis at 19 weeks gestation. J Ultrasound Med 1991; 10(2): 101-5.
10. Bromley B, Parad R, Estroff JA, Benacerraf BR. Fetal lung masses: prenatal course and outcome. J Ultrasound Med 1995; 14(12): 927-36; quiz p1378.
11. Stocker JT, Madewell JE, Drake RM. Congenital cystic adenomatoid malformation of the lung. Hum Pathol 1977; 8(2): 155-71.
12. Bruner JP, Jarnagin BK, Reinisch L. Fetal congenital cystic adenomatoid malformation of the lung. Fetal Diagn Ther 2000; 15(6): 359-63.
13. Thorpe-Beeston JG, Nicolaides KH. Cystic adenomatoid malformation of the lung: prenatal diagnosis and outcome. Prenat Diagn 1994 ; 14(8): 677-88.
14. Dommergues M, Louis-Sylvestre C, Mandelbrot L, Aubry MC, Revillon Y, Jarreau PH, et al. Congenital adenomatoid malformation of the lung: when is active fetal therapy indicated? Am J Obstet Gynecol 1997; 177(4): 953-8.
15. MacGillivray TE, Harrison MR, Goldstein RB, Adzick NS. Disappearing fetal lung lesions. J Pediatr Surg 1993; 28(10): 1321-4; discussion 1324-5.
16. Monni G, Paladini D, Ibba RM, Teodoro A, Zoppi MA, Lamberti A, et al. Prenatal ultrasound diagnosis of congenital cystic adenomatoid malformation of the lung: a report of 26 cases and review of the literature. Ultrasound Obstet Gynecol 2000; 16(2): 159-62.
17. Cha I, Adzick NS, Harrison MR, Finkbeiner WE. Fetal congenital cystic adenomatoid malformation of the lung: a clinicopathologic study of eleven cases. Am J Surg Pathol 1997; 21(5): 537-44.
18. Sheffield EA, Addis BJ, Corrin B, McCabe MM. Epithelial hyperplasia and malignant change in congenital lung cysts. J Clin Pathol 1987; 40(6): 612-4.
19. Ueda K, Gruppo R, Unger F, Martin L, Bove K. Rhabdomyosarcoma of lung arising in congenital cystic adenomatoid malformation. Cancer 1977; 40(1): 383-8.
20. Monni G, Paladini D, Ibba RM, Teodoro A, Zoppi MA, Lamberti A, et al. Prenatal ultrasound diagnosis of congenital cyst adenomatoid malformation of the lung: a report of 26 cases and review of the literature. Ultrasound Obstet Gynecol 2000; 16(2): 159-62.
21. Clark SL, Vitale DJ, Minton SD, Stoddard RA, Sabey PL. Successful fetal therapy for cystic adenomatoid malformation associated on the second-trimester hydrops. Am J Obstet Gynecol 1987; 157(2): 294-5.
22. Nugent CE, Hayashi RH, Rubin J. Prenatal treatment of type I congenital cystic adenomatoid malformation by intrauterine fetal thoracentesis. J Clin Ultrasound 1989; 17(9): 675-7.
23. Nicolaides KH, Blott M, Greenough A. Chronic drainage of fetal pulmonary cyst. Lancet 1987; 1(8533): 618.
24. Adzick NS, Harrison MR, Crombleholme TM, Flake AW, Howell LJ. Fetal lung lesions: management and outcome. Am J Obstet Gynecol 1998 ; 179(4): 884-9.
25. Mandelbrot L, Domergues MA, Aubry MS. Formations adenomatoides du poumon: facteurs pronostiques et indications thérapeutiques. Médicine Fetale 1992; 11: 15-6.
26. Stocker TJ, Madewell JE, Drake RM. Congenital cystic adenomatoid malformation of the lung: classification and morphologic spectrum. Hum Pathol 1977; 8(2): 155-71.
27. Adzick NS. The Fetus with a Lung Mass. In: Harrison MR, Evans MI, Adzick NS. The unborn patient. Philadelphia: WB Saunders, 2001: 287-96.
28. Filler R, Forte V. Respiratory problem related to airway and luna. In: O’Neill J, Rowe M, Grosfeld J, Fonkalsrud E, Coran A, eds. Pediatric Surgery. 5th ed. Baltimore: Mosby, 1998: 873-97.

