










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkRevista Médica del Uruguay
versión On-line ISSN 1688-0390
Rev. Méd. Urug. vol.21 no.2 Montevideo jun. 2005
Retardo mental, malformaciones congénitas y aberraciones cromosómicas subteloméricas crípticas
Dr. Máximo E. Drets*, Br. Federico F. Santiñaque†
Servicio Nacional de Clasificación Celular y Citometría de Flujo.
Instituto de Investigaciones Biológicas Clemente Estable
Resumen
Los recientes progresos tecnológicos ocurridos en citogenética molecular han permitido la detección de numerosas minúsculas aberraciones en la región cromosómica contigua al telómero o segmento subtelomérico, que han sido relacionadas con diversos cuadros de retardo mental, malformaciones congénitas y otros síndromes de interés médico. Investigaciones previas, llevadas a cabo mediante exploración microfotométrica y análisis gráfico computacional del subtelómero, revelaron diferentes patrones de distribución de las densidades de la cromatina y la existencia de intercambios sumamente pequeños entre cromátidas hermanas en dicho segmento cromosómico. La detección de aberraciones crípticas sindromáticas, el elevado número de intercambios cromosómicos y las observaciones microscópicas sobre la estructura subtelomérica sugieren que reflejan la gran actividad genómica y la complejidad estructural prevalente en la región. En la presente revisión se describen brevemente varios síndromes cromosómicos subteloméricos, así como la estructura molecular y citológica de la región subtelomérica y las principales funciones del segmento cromosómico terminal a fin de brindar un panorama general sobre esta área de investigaciones en rápido crecimiento de considerable significación biomédica.
Palabras clave: RETARDO MENTAL Y MALFORMACIONES CONGÉNITAS.
ABERRACIONES CROMOSÓMICAS CRÍPTICAS.
* Investigador Emérito, Instituto de Investigaciones Biológicas Clemente Estable (IIBCE), Servicio Nacional de Clasificación Celular y Citometría de Flujo.
† Becario del Programa de Desarrollo de Ciencias Básicas (PEDECIBA), IIBCE, Unidad Asociada de la Facultad de Ciencias.
Correspondencia: Dr. Máximo E. Drets
Av. Italia 3318, CP 11600, Montevideo, Uruguay.
E-mail: drets@chasque.apc.org
Recibido: 2/12/04.
Aceptado: 28/1/05.
Introducción
La región subtelomérica es un peculiar segmento multifuncional localizado contiguo a la región terminal del cromosoma o telómero, el cual está formado por ácido desoxirribonucleico (ADN) repetido y componentes proteicos específicos que recubren al extremo cromosómico y mantienen constante su integridad estructural y funcional.
El reciente hallazgo en esta región de un considerable número de aberraciones cromosómicas relacionadas con síndromes de retardo mental y malformaciones congénitas ha incrementado extraordinariamente los estudios destinados al esclarecimiento de su estructura y función en el genoma humano, habiéndose tornado en un explosivo campo de investigaciones y publicaciones por sus múltiples potenciales aplicaciones en patología y otros problemas biológicos generales.
El objetivo de la presente revisión es brindar una introducción general acerca de esta nueva área, de tan extensa proyección clínica, haciendo especial referencia a los recientes avances acaecidos tanto a nivel microscópico como molecular sobre el telómero y su región contigua, el subtelómero, que puedan resultar de interés para el médico y el biólogo. Se abordan, por tanto, en forma resumida un número de tópicos relacionados con la estructura y función de dichos segmentos cromosómicos con el propósito de suministrar una visión actualizada sobre los papeles que desempeñan durante los procesos de replicación celular en el cáncer, en el envejecimiento celular y en la producción de síndromes de retardo mental y malformaciones congénitas últimamente descubiertos.
Retardo mental, malformaciones congénitas y aberraciones cromosómicas
El capítulo de la asociación sindromática de retardo mental y malformación congénita comenzó a dilucidarse citogenéticamente cuando Lejeune y colaboradores(1) descubrieron la trisomía 21, y Sutherland(2) halló que la fragilidad del cromosoma sexual X se podía detectar mediante cultivo celular en medios de cultivo carentes de ácido fólico.
Estas aberraciones cromosómicas determinantes de los síndromes de Down y de Martin-Bell, respectivamente, son las causas más frecuentes de retardo mental y malformación congénita. Sin embargo, un importante número de portadores de retardo mental y malformaciones congénitas continuaron siendo cuadros de origen inexplicable, por lo que fueron catalogados como "idiopáticos". Sólo recientemente ha sido posible establecer su origen citogenético debido a los perfeccionamientos citológicos y moleculares que posibilitaron la detección de aberraciones mínimas producidas en la región subtelomérica no advertidas previamente por los métodos citológicos convencionales, por lo que que han sido denominadas "crípticas", siendo responsables de por lo menos 7,5% de los casos de retardo mental idiopático(3). Se sostiene que las duplicaciones o delecciones producidas en el ADN de esa región cromosómica menores de 5Mb causan retardo mental sindrómico(4) lo que señala que aun muy pequeñas anomalías génicas son capaces de originar graves perturbaciones, en particular casos inexplicables de retardo mental(5).
La tabla 1 presenta una lista parcial sobre los variados tipos de aberraciones cromosómicas detectadas en los últimos años en la región subtelomérica. Un examen detenido de la tabla evidencia el hecho de que no se ha logrado, hasta el presente, establecer algún cuadro clínico definido asociado a una aberración cromosómica específica, sino que se ha observado una compleja constelación de delecciones, duplicaciones, translocaciones e inversiones, inclusive mosaicos celulares(6,7) que afectan diversos pares cromosómicos pero que, cada una de ellas, son capaces de originar, en mayor o menor grado, retardo mental de variada intensidad y malformaciones congénitas. Sin duda, no deja de sorprender que existan tan diversas aberraciones(8) producidas en una pequeña región cromosómica como lo es el segmento subtelomérico, por lo que las complicadas fórmulas de aberraciones reseñadas someramente en la tabla 1, y muchas otras no citadas, representan un verdadero reto diagnóstico aun para un avezado citogenetista. Se aguarda plausiblemente que la reiterada aplicación de las nuevas técnicas citogenéticas permitirá descubrir muchos más micro-reordenamientos intersticiales aumentando de este modo la complejidad diagnóstica de esta nueva área citológica(9).
En general los criterios que se emplean para indicar un estudio citogenético de la región subtelomérica se basan en: a) historia clínica con dos o más individuos afectados de retardo mental; b) retardo del desarrollo prenatal; c) anomalías del crecimiento posnatal; d) dismorfia facial; e) anomalías congénitas sin dismorfia facial; y f) cariotipo normal con bandeo G. La clínica de las constituciones genéticas anormales originadas por aberraciones cromosómicas subteloméricas que se han relacionado con los síndromes de retardo mental y malformación se ha tornado tan extensa y compleja que cualquier intento de describirla en forma detallada escapa completamente a los propósitos de esta breve revisión, por lo que se sugiere al lector consultar referencias que comprendan estudios amplios sobre el problema(10-15).
Debido a la creciente importancia médica de esta región cromosómica resulta de interés, por tanto, describir las características citológicas y la constitución molecular de la región subtelomérica y sus vínculos con su segmento cromosómico contiguo, el telómero, de modo de proporcionar al clínico interesado los fundamentos generales que le permitan comprender más detalladamente los procesos citogenéticos que allí ocurren y que se relacionan con la patología congénita mencionada.
Estructura general del cromosoma eucariótico
Los cromosomas son los vehículos del material hereditario y, salvo algunas excepciones, sólo se pueden observar mediante el microscopio óptico o el electrónico cuando la célula se está dividiendo, estadio en el cual los cromosomas alcanzan su grado máximo de compactación.
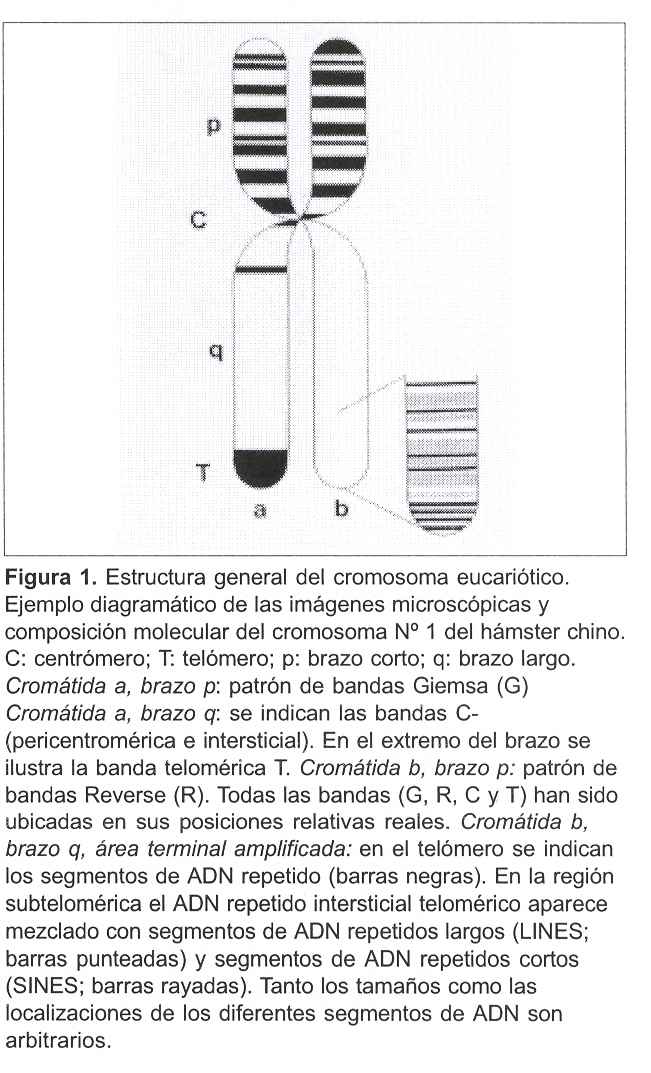
Un cromosoma metafásico típico (figura 1) está constituido por dos cromátidas hermanas, las cuales permanecen unidas mediante proteínas de adhesividad principalmente a nivel del centrómero(42). Este dominio estructural es fundamental en la producción de la correcta segregación cromosómica y, por tanto, de la información genética a las células hijas. Cada cromátida posee en su extremo una compleja región denominada telómero, la cual permite la existencia y el mantenimiento de los cromosomas lineales a lo largo de la evolución de los seres vivos(43).
Bandas cromosómicas y su importancia en la práctica médica
El cromosoma eucariótico se caracteriza por presentar estructuras transversales denominadas bandas cromosómicas. Las bandas se revelan mediante el empleo de varios métodos y técnicas de tinción. Según el patrón de bandas obtenido, las técnicas de bandeo cromosómico se clasifican en: a) técnicas de bandeo diferencial (bandeo G, Q y R) y, b) técnicas de bandeo selectivo (bandeo T y C)(44). En la tabla 2 se resumen las características más importantes de los principales patrones de bandeo.
Las bandas cromosómicas permiten la exacta identificación de cada par cromosómico, así como el diagnóstico de aneuploidías determinantes de síndromes congénitos. Se utilizan, además, como patrón de referencia para el mapeo de genes y oncogenes humanos, lo que resulta sumamente útil en la descripción citogenética de cuadros clínicos portadores de problemas hereditarios, habiéndose tornado asimismo en una herramienta esencial para el diagnóstico diferencial de un gran número de síndromes constitucionales(45).
Organización y compactación del ADN en el cromosoma metafásico
El ADN cromosómico está compuesto por secuencias nucleotídicas altamente repetidas, medianamente repetidas y de secuencia única, siendo estas últimas generalmente secuencias génicas codificantes o reguladoras. Las secuencias altamente repetidas se localizan en los cromosomas humanos principalmente en las regiones pericentroméricas heterocromáticas que se pueden observar al microscopio como bandas C(46).
Las secuencias medianamente repetidas pueden estar dispuestas en tándem, como es el caso de los minisatélites, microsatélites y del ADN telomérico, o dispersas a lo largo del cromosoma en forma de secuencias dispersas cortas (SINEs; "short interspersed elements") y de secuencias dispersas largas (LINEs; "long interspersed elements"). Tanto en los cromosomas del ratón, hámster chino, como humanos, las secuencias SINEs se encuentran concentradas en las bandas R, mientras que las secuencias LINEs se agrupan principalmente en las bandas G (tabla 2)(47-49) .
El cromosoma eucariótico es una estructura molecular altamente organizada, cuya molécula de ADN interactúa con una amplia gama de proteínas, en particular las histonas, las cuales constituyen una familia de proteínas básicas, altamente conservadas a lo largo de la evolución de las especies.
Las histonas intervienen en la producción de un primer nivel de compactación de la cromatina mediante la formación de un octámero proteico denominado nucleosoma(50). La molécula de ADN realiza dos giros alrededor de cada uno de dichos octámeros, formando una estructura denominada fibra nucleosómica. A su vez, esta fibra alcanza un segundo nivel de compactación por la interacción de proteínas adicionales resultando en un enrollamiento helicoidal para formar la denominada fibra de 30 nm o estructura en solenoide(51). No se conoce completamente cómo se compactan después estas fibras para formar el cromosoma metafásico. Al respecto, se supone que la fibra de 30 nm forma bucles que están anclados a un "andamiaje" cromosómico, el cual determina la morfología característica del cromosoma metafásico. Dicho "andamiaje" es observable mediante microscopía electrónica en cromosomas en los que se han extraído las histonas, habiéndose hallado que está plegado en forma helicoidal en las regiones de las bandas G(52).
El telómero y la región subtelomérica. Componentes estructurales y funcionales
El término telómero (del griego telos, extremo y mero, partícula) fue acuñado por Hans Muller (Premio Nobel, 1946) para designar la porción terminal del cromosoma en 1940(53). Muller advirtió, durante sus investigaciones sobre la frecuencia y tipos de reordenamientos cromosómicos producidos por los rayos X, que la producción de aberraciones terminales era muy rara, por lo que dedujo que esa área debía poseer una función de protección para el cromosoma. Es admirable comprobar que en una época tan lejana, cuando ni se sospechaba que el ADN era la molécula clave que comandaba genéticamente a la célula, Muller haya tenido la exacta percepción de imaginar que debía existir, en los extremos cromosómicos, una estructura especializada destinada a preservar su integridad estructural. Los telómeros se descubrieron por primera vez, como entidades cromosómicas diferenciadas, en protozoarios ciliados tales como Tetrahymena y Oxytricha(54,55), sobre las cuales se han realizado numerosos estudios.
Estas regiones cromosómicas permiten a la célula distinguir entre cromosomas intactos y fracturados, y su pérdida origina cambios cromosómicos y celulares irreversibles que pueden originar neoplasias o muerte celular programada (apoptosis)(56,57). A su vez, esta región cromosómica está involucrada en los mecanismos de regulación de la expresión de genes adyacentes, en el inicio del apareamiento de cromosomas homólogos durante la meiosis, en el envejecimiento celular y en la carcinogénesis(58).
Los extremos cromosómicos poseen una composición y organización particular de la cromatina, lo cual posibilita su tinción selectiva mediante la técnica de bandeo T (figura 1), que revela el segmento donde se localizan los telómeros y la región subtelomérica. Esta técnica deriva del método citológico para producir bandeo R, ambos procedimientos desarrollados por Dutrillaux(59), y se basa en la incubación de las preparaciones cromosómicas durante un período determinado de tiempo, en un buffer fosfato-ácido cítrico a 87º C, seguido de tinción con Giemsa. Las bandas T son regiones muy resistentes a la desnaturalización por calor(60) con un una riqueza relativa en pares GC, y pueden ser, a su vez, muy ricas en secuencias Alu (SINEs). Con este simple método se tiñen, en forma diferencial, los segmentos terminales cromosómicos, los cuales abarcan la región subtelomérica por lo que no es, evidentemente, un método para revelar a los telómeros ya que, como lo señalaremos más adelante, son estructuras moleculares no visualizables con el microscopio óptico.
Mediante microscopía electrónica el segmento subtelomérico aparece como una apretada madeja de filamentos(61), que refleja la compleja asociación subyacente del ADN con las proteínas características de la región, lo cual le confiere una elevada resistencia a la digestión por endonucleasas(62).
Los progresos ocurridos en los últimos años sobre el conocimiento de estas regiones cromosómicas han determinado que el telómero se haya tornado en un concepto estrictamente molecular y que las observaciones microscópicas que se refieren a las complejas estructuras término-subterminales corresponden a un nivel organizacional superior, en el cual se observan las aberraciones cromosómicas que se relacionan con diversos cuadros patológicos en el ser humano.
ADN telomérico
En la mayoría de los organismos eucariotas los telómeros están constituidos por la repetición en tándem de una secuencia muy corta de ADN, generalmente rica en los nucleótidos guanina (G) y citosina (C)(63-65). En los cromosomas humanos la secuencia telomérica es TTAGGG(66) (figura 1), la cual está altamente conservada en los vertebrados(67) y abarca una extensión de unos 5 a 15 kb. Esta secuencia se ha encontrado también en regiones pericentroméricas de cromosomas de algunos mamíferos así como dispersa en sitios intersticiales del genoma(68).
El extremo físico del ADN telomérico culmina en una larga monohebra rica en G, generando un extremo 3’ saliente, el cual se ha observado en todos los extremos cromosómicos, lo que sugiere que debe existir un procesamiento nucleolítico de la hebra rica en C(69) . El extremo 3’ saliente del ADN telomérico está muy conservado en los mamíferos y otros eucariotas, y se cree que tiene relación con la formación y estabilización de una estructura telomérica especializada denominada bucle-t (t-loop).
El modelo del bucle-t supone que la región terminal de la molécula de ADN forma un gran bucle terminal, que incluye el extremo 3’ saliente en el ADN telomérico de doble cadena(70). Este modelo se basó en observaciones de microscopía electrónica de fibras cromatínicas teloméricas que aparecen in vitro como bucles terminales cerrados en forma de bucle-t, habiéndose aislado estos bucles en células humanas y de ratón, lo que sugiere que muy probablemente son estructuras presentes in vivo(71).
ADN subtelomérico
La región subtelomérica es extraordinariamente compleja y está formada por una mezcla de regiones de ADN de secuencia única con distintas familias de ADN medianamente repetido que se caracterizan por ser polimórficas(58).
La región subtelomérica está organizada, en general, en dos áreas o subdominios, uno distal con respecto al centrómero (figura 1), caracterizado por la presencia de secuencias repetidas cortas que son comunes a varios cromosomas, y otro proximal con secuencias repetidas largas comunes a unos pocos cromosomas. Estos dos subdominios estarían separados entre sí por una región de repetidos teloméricos (TTAGGG)n(72). Como resultado del mapeo genómico y análisis inicial de las secuencias subteloméricas de casi todos los cromosomas humanos(73), se ha determinado que la región subtelomérica está enriquecida 25 veces en secuencias teloméricas, las cuales forman islas de (TTAGGG)n con un tamaño promedio de 151-200 pb, pudiendo estar agrupadas o dispersas, tanto en el ADN repetido como en el de secuencia única (figura 1). Debe destacarse que la región subtelomérica posee una concentración de genes más elevada que el resto del genoma donde, además, se encuentra el mayor número de oncogenes mapeados hasta el presente(72,73).
Los segmentos subteloméricos son regiones extraordinariamente dinámicas, variables y de rápida evolución en el genoma humano(74) determinando que sea una fuente potencial de diversidad fenotípica pero, al mismo tiempo, representando un lugar de reordenamientos cromosómicos que puede derivar en enfermedades congénitas(75).
Proteínas teloméricas
Los telómeros son un complejo nucleoproteico que proporciona a los cromosomas un recubrimiento protector, asegurando su estabilidad y la correcta segregación de la información genética durante la división celular, manteniendo la estructura cariológica característica de cada especie (figura 2)(76). Dicho "recubrimiento telomérico" está asegurado por la compleja estructura formada por las proteínas y el ADN telomérico existente en el extremo cromosómico(77).
Se han hallado hasta el presente tres proteínas que son capaces de reconocer y unirse específicamente a las regiones de ADN telomérico, es decir, a las regiones de repetición en tándem de la secuencia TTAGGG. Estas proteínas son TRF1(78), TRF2 y POT1. Las primeras dos reconocen exclusivamente al ADN bifilar, mientras que POT1 sólo reconoce a la monohebra de ADN rica en G. A su vez, se han caracterizado varias proteínas que si bien no reconocen específicamente la secuencia telomérica, forman parte de los telómeros integrando grandes complejos proteicos con TRF1 o TRF2. La proteína TRF2 es una de las proteínas más importantes en la formación del bucle-t y, al igual que TRF1, interviene en la estabilización de dicha estructura(79,80).
Métodos de detección de aberraciones subteloméricas crípticas
Las aberraciones cromosómicas de tamaño menor a cinco megabases no son observables por las técnicas de bandeo convencionales, ya que ellas no alteran el patrón de bandas obtenido por lo que, en citogenética, se denominan "crípticas". A fin de poder detectar las aberraciones subteloméricas, se pueden emplear: 1) marcadores subteloméricos polimórficos que evidencian desviaciones de la herencia mendeliana de alelos de sitios cromosómicos polimórficos cercanos a los telómeros(81,82); 2) métodos citogenéticos moleculares, basados en: a) técnicas de hibridación genómica comparativa (CGH; Comparative Genomic Hybridization)(83) o, b) la técnica de hibridización in situ fluorescente (FISH; Fluorescence in situ hibridization)(84). La técnica CGH permite la comparación de una muestra de ADN de una persona afectada con la de un individuo normal. En este método ambas muestras son fragmentadas y marcadas con distintos fluorocromos, para posteriormente mezclarlas e hibridarlas a una metafase normal. El análisis de los perfiles de fluorescencia de cada fluorocromo puede evidenciar alteraciones tales como delecciones o amplificaciones génicas en cada cromosoma.
El método FISH posibilita la localización precisa de cualquier secuencia de ADN en el cromosoma, ya que utiliza una monohebra de ADN específica de dicha región, denominada sonda, la cual se conjuga con uno o varios fluorocromos y reconoce e hibrida únicamente a la secuencia complementaria del ADN cromosómico previamente desnaturalizado(85). Esta técnica tiene la ventaja de que puede utilizarse tanto en cromosomas como en núcleos interfásicos(86). En los últimos años se han incrementado las investigaciones destinadas a obtener métodos diagnósticos cada vez más precisos y confiables, disponiéndose actualmente de sondas subteloméricas para segmentos muy próximos a los telómeros de casi todos los cromosomas(87), conjugadas con distintas combinaciones de fluorocromos, específicas para cada par cromosómico. Como se emplean sistemas computacionales apropiados, se logra una rápida detección de dichas aberraciones crípticas facilitando considerablemente el diagnóstico diferencial en clínica(88).
Telomerasa, envejecimiento celular y cáncer
El mantenimiento de la integridad y estabilidad de los extremos cromosómicos a través de las sucesivas replicaciones celulares está íntimamente vinculado a la longitud del ADN telomérico, la cual se establece durante el desarrollo del cigoto(89,90). Aunque parezca paradójico, esta estructura tan importante para la estabilidad cromosómica se degrada sucesivamente en cada ciclo de replicación del ADN. Esto es, en parte, debido a la incapacidad replicativa para completar el extremo terminal de la hebra de ADN retrasada y también por la acción de nucleasas, que intervienen en la generación de los extremos 3’ salientes(91,69). El acortamiento progresivo del ADN telomérico actúa como un verdadero reloj biológico(92), ya que el momento en que una célula envejece y muere depende del número de divisiones celulares previas. Este fenómeno, denominado "senescencia celular", explica por qué mueren las líneas celulares en cultivo después de cierto número de pasajes in vitro(93).
Dicho acortamiento telomérico es observado en la mayoría de las células somáticas del organismo adulto, pero no en la línea germinal o en las primeras etapas del desarrollo embrionario(94), debido a que en éstas el acortamiento es contrarrestado por la actividad de una enzima crítica, la telomerasa(95) (figura 3). La enzima telomerasa es una transcriptasa reversa específica del telómero que extiende el ADN repetido terminal (TTAGGG) proporcionando, prácticamente en forma universal, las bases moleculares para un ilimitado potencial de proliferación celular.
La telomerasa está constituida por una subunidad catalítica proteica y por una molécula de ARN, la cual posee la secuencia complementaria de los repetidos teloméricos (AAUCCC)(94). De esta forma, la telomerasa le adiciona al ADN terminal secuencias repetidas teloméricas, utilizando su propio ARN como molde. La actividad de la telomerasa está ausente en la mayoría de las células somáticas humanas, pero presente en varias líneas celulares inmortalizadas in vitro y en 90% de las células cancerosas, en las cuales se reactiva dicha enzima, por lo que se interrumpe en ellas la declinación del acortamiento telomérico(96,97). Si bien se cree que la activación de esta enzima no es responsable de la transformación celular maligna, ella permite la proliferación indefinida de las células neoplásicas(98). No obstante, se observa en casi todos los tumores que la longitud telomérica se mantiene finalmente en forma estable, lo que sugiere que los mecanismos de regulación existentes en la región limitan, en ciertos casos, el alargamiento telomérico indefinido producido por la telomerasa(76,99).
Indicaciones sobre la complejidad citológica de la región subtelomérica
Las regiones subteloméricas se pueden analizar mediante un sistema de microscopio fotométrico asociado a un sistema de computadora gráfica. Aparte de detalles técnicos menores, el microfotómetro consta básicamente de una electroplatina para microscopio que se mueve por pasos discretos (0,25 µm) y una fotocélula de alta sensibilidad (fotomultiplicadora), funcionando ambos dispositivos bajo comando computacional. Un programa apropiado determina que la platina se desplace paso a paso realizando, en cada uno, medidas muy precisas de la luz transmitida por el objeto analizado. Los valores medidos de las diferentes densidades son almacenados en la computadora que se emplean, en tiempo diferido, para generar imágenes gráficas de las estructuras cromosómicas. Tanto los programas computacionales diseñados por nosotros para analizar las estructuras nucleares y cromosómicas, como las imágenes generadas, han sido presentados detalladamente en diversos trabajos previos(100). El resultado final es un valioso sistema de microscopía analítica ya que el investigador dispone de una imagen cuantitativa muy ampliada y detallada de estructuras cromosómicas o de otros componentes celulares.
La exploración microfotométrica de las regiones terminales de cromosomas teñidos con el procedimiento de bandeo T y el posterior análisis computacional de las imágenes gráficas generadas permite detectar que las áreas de mayor densidad teñidas con el Giemsa se distribuyen de manera diferencial en ambas cromátidas hermanas. Sucintamente, son: 1) de igual tamaño en las dos cromátidas, 2) mayor en una que en la otra, o 3) sólo detectables en una única cromátida. Ambas estructuras diferenciales no pudieron asignarse a ningún cromosoma en particular(101). La existencia de estas inesperadas estructuras fue confirmada posteriormente analizando cromosomas endorreduplicados ya que en ambos cromosomas el patrón de distribución fue exactamente igual(102) (figura 4).
Un análisis detenido de las imágenes gráficas microfotométricas muestra que, en ciertos casos, las densidades más elevadas de la cromatina se distribuyen de tal modo que aparentan intercambios de cromátidas hermanas muy pequeños ocurridos en la región subtelomérica, los cuales hemos denominado "t-SCEs" (de: intercambios de cromátidas hermanas teloméricos)(101). Como este método de análisis no implica la sustitución de bases del ADN con bromodeoxiuridina (BrdU), que es el método citológico empleado corrientemente para detectar los intercambios de cromátidas hermanas, nosotros postulamos que este era un fenómeno que representa distintos estadios funcionales de la región del cromosoma normal(103).
Mediante microfotometría de exploración hemos establecido también que tanto las señales fluorescentes generadas por las sondas teloméricas como las regiones de mayor densidad de la cromatina teñidas por el método de bandeo T se localizan prácticamente en las mismas áreas en la región subtelomérica, lo que sugiere que ambos hechos están relacionados por lo menos a nivel del microscopio óptico(104). Otro singular hallazgo sobre la reactividad subtelomérica fue la induccción de pequeños orificios en dicha región tanto en cromosomas normales como aberrantes y en ambas cromátidas o en una sola, cuyas localizaciones evocan la de los diferentes patrones de densidades mencionados(105,106).
Un hecho semejante a la distribución diferencial de las densidades cromatínicas fue observado también por Bekaert y colaboradores(107) estudiando el tamaño del telómero en tercera dimensión mediante microscopía confocal y sondas fluorescentes teloméricas, comprobaron que la longitud telomérica no era igual en las dos cromátidas hermanas de cromosomas de linfocitos humanos y que esta variabilidad no podía relacionarse con algún par cromosómico específico, lo que concuerda con nuestras observaciones sobre la distribución de los patrones diferenciales de densidades.
Discusión y conclusiones
La citogenética humana ha realizado nuevamente un aporte clave al dilucidar la causa de múltiples cuadros de retardo mental y malformaciones congénitas asociadas, un antiguo enigma de la patología humana. Debido a estos importantes avances acaecidos en esta área, el clínico actual no sólo debe familiarizarse con este nuevo conjunto de síndromes para realizar un diagnóstico diferencial preciso, sino que también debe disponer de los fundamentos conceptuales subyacentes en estas alteraciones congénitas.
En esta sucinta revisión se presentan, por tanto, una descripción citogenética general de algunos tipos de aberraciones observadas en dichos síndromes así como aquellos aspectos estructurales y moleculares del telómero y la región subtelomérica que se relacionan con la producción de aberraciones cromosómicas que originan retardo mental y malformación congénita a fin de brindar al médico una visión general sobre el problema.
El descubrimiento de estas aberraciones crípticas ha sido posible por los recientes refinamientos y nuevos métodos citogenéticos de análisis cromosómico, en particular el diseño de sondas fluorescentes desarrolladas para la región subtelomérica. Hasta hace muy poco era inimaginable que la alteración de pequeñas porciones de los cromosomas se tornaría en áreas críticas en patología humana. La multiplicidad de hallazgos es ahora de tal magnitud que se sostiene que por lo menos 6% del retardo mental "idiopático" podría explicarse por los reordenamientos submicroscópicos que afectan la región subtelomérica(87).
El hecho de que la región subtelomérica sea el sitio de un elevado número de intercambios, un hecho comprobado durante la decodificación del genoma de todos los cromosomas humanos(108,109,73), permite comprender por qué se están detectando en forma creciente numerosas aberraciones "crípticas" capaces, aparentemente, de generar variabilidad fenotípica, síndromes de retardo mental y perturbaciones del desarrollo embrionario.
Resulta realmente sorprendente apreciar cómo tan pequeñas alteraciones cromosómicas, cuyo tamaño está casi en el límite de resolución del microscopio, sean capaces de originar complejos cuadros clínicos de retardo mental y malformación congénita. Probablemente dichas aberraciones provocan un considerable desequilibrio génico que perturba el desarrollo normal del embrión, lo que originaría dichos síndromes.
En este sentido, Pettenati y colaboradores(110) hallaron que pueden ocurrir reordenamientos cromosómicos entre la región subtelomérica y telomérica en individuos clínicamente normales que portan anomalías cromosómicas equilibradas en las cuales uno de los puntos de fractura involucra una banda terminal. Como estas regiones intervienen durante el apareamiento cromosómico meiótico, dichas alteraciones perturbarían la formación de las configuraciones cromosómicas normales, lo que interferiría con la producción gamética originando abortos e individuos portadores de aberraciones cromosómicas.
La figura 4 resume, en forma diagramática, los hechos principales registrados hasta la fecha sobre esta área. La comprobada complejidad de ambas estructuras es aún materia de controversia. Persisten así una serie de interrogantes sobre cómo está organizada la cromatina en el cromosoma metafásico y cómo funciona y está estructurado realmente el conjunto molecular. Debido a esto es anticipable que se incrementarán de manera considerable las investigaciones sobre este segmento del cromosoma, en particular a nivel molecular. Asimismo, a medida que se desarrollen nuevas sondas específicas conjugadas con fluorocromos de alta eficiencia cuántica, se logrará detectar un número mayor de síndromes subteloméricos de interés clínico. Además, es evidente que en un futuro próximo se asignarán a la región muchas otras funciones aún desconocidas, considerando la elevada concentración génica predominante en la región característica de las bandas R y T, por lo que se obtendrá una imagen más precisa sobre estas áreas cromosómicas.
Aunque la microscopía electrónica permite visualizar la complejidad estructural existente en el segmento subtelomérico y que el análisis microfotométrico muestra la existencia de variabilidad en la distribución de las densidades de la cromatina, todavía no se ha proporcionado una imagen global convincente sobre cómo están asociados el ADN y las proteínas en esa región que explique satisfactoriamente las estructuras observadas a nivel microscópico, las cuales parecen poseer cierta impenetrabilidad estructural.
Tanto los diferentes patrones de densidad detectados en la región subtelomérica por nosotros, como las distintas longitudes teloméricas halladas por Bekaert y colaboradores(107) son hechos no aguardables de un punto de vista citogenético estrictamente ortodoxo dado que las cromátidas hermanas normales no debieran exhibir diferencias estructurales debido a que cada una está formada por una única molécula de ADN y, en mitosis, éstas poseen exactamente la misma secuencia, ya que son el producto de la replicación semiconservativa de una única molécula lineal de ADN antecesora. Por tanto, nosotros adelantamos la hipótesis de que, probablemente, las estructuras que observamos en este segmento expresan diversos estados funcionales de la región subtelomérica, una interpretación que es coherente con las más recientes observaciones estructurales y funcionales halladas en el segmento(104). Asimismo, los pequeños intercambios que hemos detectado en las regiones subteloméricas de las cromátidas hermanas (t-SCEs) deben interpretarse también como expresiones de la activa funcionalidad del segmento. Al respecto, se ha señalado la existencia en el genoma humano de inestabilidades en las secuencias intersticiales teloméricas(111), por lo que nuestras observaciones microscópicas podrían representar imágenes de esas inestabilidades. Aunque esta idea resulta atractiva será necesario llevar a cabo extensas investigaciones sobre los cambios estructurales originados por la actividad de los diversos mecanismos que operan en esta región del cromosoma, un asunto que originará seguramente el descubrimiento de nuevos e inesperados hechos relacionados. Otro aspecto, no menos importante, será poder establecer un vínculo convincente entre los componentes moleculares y las macroestructuras detectadas a nivel microscópico. El logro de esta síntesis seguramente permitirá relacionar dichas estructuras con las diferentes funciones conocidas y otras ignoradas hasta ahora, desempeñadas por el segmento subtelomérico brindando una imagen integral de la región.
Aun cuando en la presente revisión nos hemos ocupado preferencialmente de las aberraciones crípticas subteloméricas que originan retardo mental y malformaciones congénitas en pacientes y en familias, debe señalarse que también se han encontrado aberraciones similares en otras áreas médicas, en particular en hemato-oncología(112,113), cancerología(114), psiquiatría(115,116), ginecología(117), sobre las cuales sólo citamos unas pocas referencias a título de ejemplos. Asimismo, se han comenzado a vincular diversas aberraciones crípticas con el oscuro síndrome de autismo(27,118-120), todo lo cual ha expandido considerablemente el interés biológico y diagnóstico existente sobre esta región cromosómica cuyos estudios están inaugurando una nueva espectacular especialidad en medicina.
Agradecimientos
Los autores desean expresar su sincero agradecimiento al Prof. Dr. Rodolfo Wettstein por la revisión y valiosas sugerencias realizadas sobre el manuscrito. Quedamos reconocidos al Dr. Gustavo A. Folle por su amable cooperación en la obtención de las imágenes digitales que aparecen en la figura 3. Financiado en parte por el Programa de Desarrollo de Ciencias Básicas (PEDECIBA), Uruguay.
Summary
Recent technological advances occurred in molecular cytogenetics have allowed to detect numerous minute aberrations in the chromosome region contiguous to the telomere or subtelomeric segment which has been related to various conditions of mental retardation and congenital malformations and to other clinical syndromes of medical interest. Previous research on the subtelomere carried out by means of scanning microphotometry and computer graphic analysis, revealed different patterns of the distribution of chromatin densities and the existence of minute exchanges between sister chromatids in this chromosomic segment. The detection of cryptic syndromatic aberrations, the high number of chromosome crossovers and the microscopical observations on the subtelomeric structure suggest that they reflect the great genomic activity and the structural complexity prevailing in the region. In the present review, several subtelomeric chromosome syndromes, the cytological and molecular structure of the subtelomeric region as well as the main functions of the chromosome terminal segment are briefly described in order to give a general panorama on this fast growing research area of considerable significance in bio-medicine.
Résumé
Les derniers progrès technologiques en cytogénétique moléculaire, ont permis de repérer de nombreuses et minuscules aberrations dans la région chromosomique du télomère ou segment subtélomérique liées au retard mental, des malformations congénitales et d’autres syndrômes d’intérèt médical. Des recherches antérieures, menées à bout au moyen d’exploration microphotométrique et d’analyse graphique par ordinateur du subtélomère, ont montré de différents paramètres de distribution des densités de la chromatine et l’existence de minuscules échanges entre chromatides soeurs dans ce segment chromosomique. La détection d’aberrations cryptiques syndromatiques, le grand nombre d’échanges chromoso-miques et les observations microscopiques de la structure subtélomérique suggèrent une grande activité génomique et la compléxité structurelle prévalente dans la région. On décrit ici brièvement plusieurs syndrômes chromoso-miques subtélomériques, ainsi que la structure moléculaire et cytologique de la région subtélomérique et les principales fonctions du segment chromosomique terminal, afin d’offrir un panorama général sur cette sujet de recherches qui s’accroit considérablement.
Bibliografía
1. Lejeune J, Gauthier M, Turpin R. Étude des chromosomes somatiques de neuf enfants mongoliens. Compt Rend Acad Sci, Paris, 1959; 248: 1721-2.
2. Sutherland GR. Fragile sites on human chromosomes: demonstration of their dependence on the type of tissue culture medium. Science 1977; 197: 265-6.
3. Sutton VR, Coveler KJ, Lalani SR, Kashork CD, Shaffer LG. Subtelomeric FISH uncovers trisomy 14p32: lessons for imprinted regions, cryptic rearrangements and variant acrocentric short arms. Am J Med Genet 2002; 112: 23-7.
4. Borck G, Rio M, Sanlaville D, Redon R, Molinari F, Bacq D, et al. Genome-wide screening using automated fluorescent genotyping to detect cryptic cytogenetic abnormalities in children with idiopathic syndromic mental retardation Clin Genet 2004; 66: 122-7.
5. Fan YS, Zhang Y, Speevak M, Farrell S, Jung JH, Siu VM. Detection of submicroscopic aberrations in patients with unexplained mental retardation by fluorescence in situ hybridization using multiple subtelomeric probes. Genet Med 2001; 3: 416-21.
6. Engelen JJ, Marcelis C, Herbergs J, Weber J, Alofs M, Albrechts JC, et al. Mosaic telomere (2;14) association in a child with motor delay. Am J Med Genet 2000; 92: 18-21.
7. Riegel M, Baumer A, Jamar M, Delbecque K, Herens C, Verloes A, et al. Submicroscopic terminal deletions and duplications in retarded patients with unclassified malformation syndromes. Hum Genet 2001; 109: 286-94.
8. Patsalis PC, Evangelidou P, Charalambous S, Sismani C. Fluorescence in situ hybridization characterization of apparently balanced translocation reveals cryptic complex chromosomal rearrangements with unexpected level of complexity. Eur J Hum Genet 2004; 12: 647-53.
9. Kriek M, White SJ, Bouma MC, Dauwerse HG, Hansson KB, Nijhuis JV, et al. Genomic imbalances in mental retardation. J Med Genet 2004; 41: 249-55.
10. Baker E, Hinton L, Callen DF, Altree M, Dobbie A, Eyre HJ, et al. Study of 250 children with idiopathic mental retardation reveals nine cryptic and diverse subtelomeric chromosome anomalies. Am J Med Genet 2002; 107: 285-93.
11. Bocian E, Helias-Rodzewicz Z, Suchenek K, Obersztyn E, Kutkowska-Kazmierczak A, Stankiewicz P, et al. Subtelomeric rearrangements: results from FISH studies in 84 families with idiophatic mental retardation. Med Sci Monit 2004; 10: 143-51.
12. de-Vries BB, White SM, Knight SJ, Regan R, Homfray T, Young ID, et al. Clinical studies on submicroscopic subtelomeric rearrangements: a checklist. J Med Genet 2001; 38: 145-50.
13. Novelli A, Ceccarini C, Bernardini L, Zuccarello D, Caputo V, Digilio MC, et al. High frequency of subtelomeric rearrangements in a cohort of 92 patients with severe mental retardation and dysmorphism. Clin Genet 2004; 66: 30-8.
14. Roberts AE, Cox GF, Kimonis V, Lamb A, Irons M. Clinical presentation of 13 patients with subtelomeric rearrangements and a review of the literature. Am J Med Genet 2004; 128: 352-63.
15. Walter S, Sandig K, Hinkel GK, Mitulla B, Ounap K, Sims G, et al. Subtelomere FISH in 50 children with mental retardation and minor anomalies, identified by a checklist, detects 10 rearrangements including a de novo balanced translocation of chromosomes 17p13.3 and 20q13.33. Am J Med Genet 2004; 128: 364-73.
16. Bartsch O, Hinkel GK, Petersen MB, Konig U, Bugge M, Mikkelsen M, et al. A large family with subtelomeric translocation t(18;21;(q23;q22.1; and molecular breakpoint in the Down syndrome critical region. Hum Genet 1997; 100: 669-75.
17. Bonaglia MC, Giorda R, Borgatti R, Felisari G, Gagliardi C, Selicorni A, et al. Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am J Hum Genet 2001; 69: 261-8.
18. Brown J, Horsley SW, Jung C, Saracoglu K, Janssen B, Brough M, et al. Identification of a subtle t(16;19;(p13.3; p13.3) in an infant with multiple congenital abnormalities using a 12-colour multiplex FISH telomere assay, M-TEL. Eur J Hum Genet 2000; 8: 903-10.
19. Cargile CB, Goh DL, Goodman BK, Chen XN, Korenberg JR, Semenza GL, et al. Molecular cytogenetic characterization of a subtle interstitial del(3)(p25.3p26.2) in a patient with deletion 3p syndrome. Am J Med Genet 2002; 109: 133-8.
20. Cotter PD, Kaffe S, Li L, Gershin IF, Hirschhorn K. Loss of subtelomeric sequence associated with a terminal inversion duplication of the short arm of chromosome 4. Am J Med Genet 2001; 102: 76-80.
21. de-Vries BB, Lees M, Knight SJ, Regan R, Corney D, Flint J, et al. Submicroscopic 8pter deletion, mild mental retardation, and behavioral problems caused by a familial t(8;20)(p23;p13) Am J Med Genet 2001; 99: 314-9.
22. Giardino D, Finelli P, Gottardi G, De-Canal G, Della-Monica M, Lonardo F, et al. Narrowing the candidate region of Albright hereditary osteodytrophy síndrome by deletion mapping in a patient with unbalanced cryptic translocation t(2;6)(q37.3;q26). Am J Med Genet 2003; 122: 261-5.
23. Holinski-Feder E, Reyniers E, Uhrig S, Golla A, Wauters J, Kroisel P, et al. Familial mental retardation syndrome ATR-16 due to an inherited cryptic subtelomeric translocation, t(3;16)(q29;p13.3);. Am J Hum Genet 2000; 66: 16-25.
24. Horn D, Neitzel H, Tonnies H, Kalscheuer V, Kunze J, Hinkel GK, et al. Familial MCA/MR syndrome due to inherited submicroscopic translocation t(18;21)(q2.11q21.3) with breakpoint at the Down syndrome crtical region. Am J Med Genet 2003 117: 236-44.
25. Horsley SW, Knight SJ, Nixon J, Huson S, Fitchett M, Boone RA, et al. Del(18p) shown to be a cryptic translocation using a multiprobe FISH assay for subtelomeric chromosome rearrangements. J Med Genet 1998; 35: 722-6.
26. Jenderny J, Poetsch M, Hoeltzenbein M, Friedrich U, Jauch A. Detection of a concomitant distal deletion in an inverted duplication of chromosome 3. Is there an overall mechanism for the origin of such duplications/deficiencies? Eur J Hum Genet 1998; 6: 439-44.
27. Keller K, Williams C, Wharton P, Paulk M, Bent-Williams A, Gray B, et al. Routine cytogenetic and FISH studies for 17p11/15q11 duplications and subtelomeric rearrangements studies in children with autism spectrum disorders. Am J Med Genet 2003; 117: 105-11.
28. Kleefstra T, van-de-Zande G, Merkx G, Mieloo H, Hoovers JM, Smeets D. Identification of an unbalanced cryptic translocation between the chromosomes 8 and 13 in two sisters with mild mental retardation accompanied by mild dysmorphic features. Eur J Hum Genet 2000; 8: 637-40.
29. Leube B, Majewski F, Drechsler M, Royer-Pokora B. Unbalanced cryptic translocation der(14)t(9;14)(q34.3; q32.33) identified by subtelomeric FISH. Clin Dysmorphol 2003; 12: 261-5.
30. Lorda-Sánchez I, López-Pajares I, Roche MC, Sanz R, Rodríguez-De-Alba M, González-González MC, et al. Cryptic 6q subtelomeric deletion associated with a paracentric inversion in a mildly retarded child. Am J Med Genet 2000; 95: 336-8.
31. Precht KS, Lese CM, Spiro RP, Huttenlocher PR, Johnston KM, Baker JC, et al. Two 22q telomere deletions serendipituosly detected by FISH. J Med Genet 1998; 35: 939-42.
32. Quigley DI, Kaiser-Rogers K, Aylsworth AS, Rao KW. Submicroscopic deletion 9(34.3) and duplication 19(p13.3): identified by subtelomere FISH probes. Am J Med Genet 2004; 125: 67-72.
33. Rauch A, Beese M, Mayatepek E, Dorr HG, Wenzel D, Reis A, et al. A novel 5q35.3 subtelomeric deletion syndrome. Am J Med Genet 2003; 121: 1-8.
34. Reddy KS, Fugate JK. A half cryptic derivative der 18;t(5;18); pat identified by M-FISH and subtelomere probes: clinical findings and review of subtelomeric rearrangements. Clin Genet 1999; 56: 328-32.
35. Schultz LN, Schmidt P, Tabor A, Bryndorf T, Christesen B, Ludsteen C. Cryptic familial t11;18;(q25;q23) incidentally detected by interphase FISH. Clin Genet 2001; 59: 279-83.
36. Souter VL, Glass IA, Chapman DB, Raff ML, Parisi MA, Opheim KE, et al. Multiple fetal anomalies associated with subtle subtelomeric chromosomal rearrangements. Ultrasound Obstet Gynecol 2003; 21: 609-15.
37. Speleman F, Callens B, Logghe K, Van-Roy N, Horsley SW, Jauch A, et al. Subtelomeric familial translocation t(2;7;(q37;q35; leading to partial trisomy 7q35®qter: molecular cytogenetic analysis and clinical phenotype in two generations. Am J Med Genet 2000; 93: 349-54.
38. van-Deutekom JC, Bakker E, Lemmers RJ, Van Der Wielen MJR, Bik E, Hofker MH, et al. Evidence for subtelomeric exchange of 3.3 kb tandemly repeated units between chromosomes 4q35 and 10q26: implications for genetic counselling and etiology of FSHD1. Hum Mol Genet 1996; 5: 1997-2003.
39. Velagaleti GV, Jalal SM, Michaelis RC, Rowe TF, Nichols JR, Lockhart LH. Molecular cytogenetic characterization of a de novo unbalanced translocation leading to trisoomy 17q25®qter and monosomy 18p11.3®pter in a girl with dysmorphic features. Clin Dysmorphol 2003; 12: 29-33.
40. Warburton P, Mohammed S, Ogilvie CM. Detection of submicroscopic subtelomeric chromosome translocations: A new case study. Am J Med Genet 2000; 91: 51-5.
41. Wong ACC, Ning Y, Flint J, Clark K, Dumanski JP, Ledbetter DH, et al. Molecular characterization of a 130-kb terminal microdeletion at 22 q in a child with mild mental retardation. Am J Hum Genet 1997; 60: 113-20.
42. Sekelsky J, Hawley RS. The bond between sisters. Cell 1995; 83: 157-60.
43. Therman E, Susman, M. Human Chromosomes Structure, Behavior and Effects. Berlín:Springer, 3rd. ed 1993; 1-376; Cromosomas Humanos. Estructura, Comportamiento y Efectos. Brasil: Soc Bras Gen, ed. (Traducción: Drets, M.E.) 1996; 1-383.
44. Holmquist GP. Chromosome bands, their chromatin flavors and their functional features. Am J Hum Genet 1992; 51: 17-37.
45. Drets ME. Una saga citogenética: El descubrimiento de los métodos de bandeo cromosómico. Significado y proyección bio-médica. Rev Med Uruguay 2002; 18: 107-21.
46. Verma RS. Molecular biology of heterochromatin. In: The genome. New York: VCH Publishers, 1990: 43-72.
47. Holmquist GP, Caston LA. Replication time of interspersed repetitive DNA sequences in hamsters. Bioch Bioph Acta 1986; 868: 164-47.
48. Korenberg JR, Rykowski MC. Human genome organization: Alu, Lines and the molecular structure of metaphase chromosome bands. Cell 1988; 53: 391-400.
49. Vizard DL, Rosenberg NL. Terminal replication of an interspersed repeated sequence of mouse DNA. Bioch Bioph Acta 1984; 782: 402-7.
50. Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosomes. Cell 1999; 98: 285-94.
51. Wolffe A. Chromatin structure. In: Chromatin. Structure and Function. 3rd ed. San Diego: Academic Press, 1998: 7-172.
52. Saitoh Y, Laemmli UK. Metaphase chromosome structure: bands arise from a differential folding path of the highly AT-rich scaffold. Cell 1994; 76: 609-22.
53. Muller HJ. The remaking of chromosomes. Collecting Net 1938; 8: 182-95.
54. Klobutcher LA, Swanton MT, Donini P, Prescott DM. All gene-sized DNA molecules in four species of hypotrichs have the same terminal sequence and an unusual 3' terminus. Proc Natl Acad Sci 1981; 78: 3015-9.
55. Oka Y, Shiota S, Nakai S, Nishida Y, Okubo S. Inverted terminal repeat sequence in the macromolecular DNA of Stylonychia pustulata. Gene 1980; 10: 301-6.
56. Ostler EL, Wallis CV, Aboalchamat B, Faragher RG. Telomerase and the cellular lifespan: implications of the aging process. J Pediatr Endocrinol Metab 2000; 13: 1467-76.
57. Pardue ML, DeBarysche PG. Telomeres and telomerase: more than the end of the line. Chromosoma 1999; 108: 73-82.
58. Henderson E. Telomere DNA structure. In: Blackburn EH, Greider CW, eds. Telomeres. New York: Cold Spring Harbor Lab Press, 1995: 11-34.
59. Dutrillaux B. Nouveau système de marquage chromosomique: Les bandes T. Chromosoma 1973; 41: 395-402.
60. Ludeña P, Sentis C, De-Cabo F, Velázquez M, Fernández-Piqueras J. Visualization of R-bands in human metaphase chromosomes by the restriction endonuclease MseI. Cytogenet Cell Genet 1991; 57: 82-6.
61. Allen TD, Jack EM, Harrison CJ. The three dimensional structure of human metaphase chromosomes determined by scanning electron microscopy. In: Adolph KW, ed. Chromosomes and chromatin. Florida: CRC Press, 1988: 51-72.
62. Steiner S, Shay JW, Wright WE. Modification of subtelomeric DNA. Mol Cell Biol 2004; 24: 4571-80.
63. Blackburn EH, Gall JG. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. J Mol Biol 1978; 120: 33-53.
64. Blackburn EH, Greider CW, eds. Telomeres. New York: Cold Spring Harbor Lab Press, 1995: 1-396.
65. Zakian VA. Telomeres: beginning to understand the end. Science 1995; 270: 1601-7.
66. Moyzis RK, Buckinham JM, Cram LS, Dani M, Deaven LL, Jones MD, et al. A highly conserved repetitive DNA sequence (TTAGGG)n present at the telomeres of human chromosomes. Proc Natl Acad Sci USA 1988; 85: 6622-6.
67. Meyne J, Ratliff Rl, Moyzis RK. Conservation of the human telomere sequence (TTAGGG)n among vertebrates. Proc Natl Acad Sci USA 1989; 89: 7049-53.
68. Meyne J, Ratliff RL, Buckingham JM, Jones MD, Wilson JS, Moyzis RK. The human telomere. In: Fredga K, Kihlman BA, Bennet MD, eds. Chromosomes Today. London: Unwin Hyman, 1990: 75-80.
69. Makarov VL, Hirose Y, Langmore JP. Long G Tails at Both Ends of Human Chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell 1997; 88: 657-66.
70. Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, et al. Mammalian telomeres end in a large duplex loop. Cell 1999; 97: 503-14.
71. Nikitina T, Woodcock CL. Closed chromatin loops at the ends of chromosomes. J Cell Biol 2004; 166: 161-5.
72. Flint J, Bates GP, Clark K, Dorman A, Willingham D, Roe BA, et al. Sequence comparison of human and yeast telomeres identifies structurally distinct subtelomeric domains. Hum Mol Genet 1997; 6: 1305-13.
73. Riethman H, Ambrosini A, Castaneda C, Finklestein J, Hu XL, Mudunuri U, et al. Mapping and initial analysis of human subtelomeric sequence assemblies. Genome Res 2004; 14: 18-28.
74. Der-Sarkissian H, Vergnaud G, Borde YM, Thomas G, Londono-Vallejo JA. Segmental polymorphism in the proterminal regions of a subset of human chromosomes. Genome Res 2002; 12: 1673-8.
75. Mefford HC, Trask BJ. The complex structure and dynamic evolution of human subtelomeres. Nat Rev Genet 2002; 3: 91-102.
76. Blackburn EH. Switching and signaling at the telomere. Cell 2001; 106: 661-73.
77. de-Lange T, Shiue L, Myers RM, Cox DR, Naylor SL, Killery AM, et al. Structure and variability of human chromosome ends. Mol Cell Biol 1990; 10: 518-27.
78. Chong L, van-Steensel B, Broccoli D, Erdjument-Bromage H, Hanish J, Tempst P, et al. A human telomeric protein. Science 1995; 270: 1663-7.
79. Stansel R, de-Lange T, Griffith JD. T-loop assembly in vitro involves binding of TRF2 near the 3' telomeric overhang. EMBO 2001; 20: 5532-40.
80. Yoshimura SH, Maruyama H, Ishikawa F, Ohki R, Takeyasu K. Molecular mechanisms of DNA end-loop formation by TRF2. Genes Cells 2004; 9: 205-18.
81. Colleaux L, Rio M, Heuertz S, Moindrault S, Turleau C, Ozilou C, et al. A novel automated strategy for screening cryptic telomeric rearrangements in children with idiopathic mental retardation. Eur J Hum Genet 2001; 9: 319-27.
82. Slavotinek A, Rosenberg M, Knight S, Gaunt L, Fergusson W, Killoran C, et al. Screening for submicroscopic chromosome rearrangements in children with idiopathic mental retardation using microsatellite markers for the chromosome telomeres. J Med Genet 1999; 36: 405-11.
83. Ness GO, Lybaek H, Houge G. Usefulness of high resolution comparative genomic hybridization (CGH) for detecting and characterizing constitutional chromosome abnormalities. Am J Med Genet 2002; 113: 125-36.
84. Jalal SM, Harwood AR, Sekhon GS, Pham-Lorentz C, Ketterling RP, Babovic-Vuksanovic D, et al. Utility of subtelomeric fluorescent DNA probes for detection of chromosome anomalies in 425 patients. Genet Med 2003; 5: 28-34.
85. Pinkel D, Sstraume T, Gray JW. Cytogenetic analysis using quantitative, high-sensitivity, fluorescence hybridization. Proc Natl Acad Sci USA 1986; 83: 2934-8.
86. Ried T, Schröck E, Ning Y, Wienberg J. Chromosome painting: a useful art. Hum Mol Genet 1998; 10: 1619-26.
87. Knight SJI, Flint J. Perfect endings: a review of subtelomeric probes and their use in clinical diagnosis. J Med Genet 2000; 37: 401-9.
88. Brown J, Saracoglu K, Uhrig S, Speicher MR, Eils R, Kearney L. Subtelomeric chromosome rearrangements are detected using an innovative 12-color FISH assay (M-TEL). Nat Med 2001; 4: 497-501.
89. Graakjaer J, Pascoe L, Der-Sarkisian H, Thomas G, Kolvraa S, Christensen K, et al. The relative lengths of individual telomeres are defined in the zigote and strictly maintained during life. Aging Cell 2004; 3: 97-102.
90. McEachern MJ, Krauskopf A, Blackburn EH. Telomeres and their control. Annu Rev Genet 2000; 34: 331-58.
91. Lingner J, Cooper JP, Cech TR. Telomerasa and DNA end replication: no longer a lagging strand problem? Science 1995; 269: 1533-4.
92. Harley CB. Telomeres and Aging. In: Blackburn EH, Greider CW, eds. Telomeres. New York: Cold Spring Harbor Lab Press, 1995: 247-63.
93. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 1965; 37: 614-36.
94. Cong Y, Wright WE, Shay JW. Human telomerase and its regulation. Microbiol Mol Biol Rev 2002; 66: 407-25.
95. Blasco MA, Gasser SM, Lingner J. Telomeres and telomerase. Genes Dev 1999; 13(18): 2353-9.
96. Greider CW. Telomerase activity, cell proliferation, and cancer. Proc Natl Acad Sci USA 1998; 95: 90-2.
97. Hiyama E, Hiyama K. Clinical utility of telomerase in cancer. Oncogene 2002; 21: 643-9.
98. Morin GB. Telomere Integrity and Cancer. J Nat Cancer Inst 1996; 88(16): 1095-6.
99. Shay JW, Wright WE. Aging. When do telomeres matter? Science 2001; 291: 839-40.
100. Drets ME, Drets GA, Queirolo PJ, Monteverde FJ. Computer graphics as a tool in cytogenetic research and education. Comp Appl Biosc (CABIOS) 1995; 11: 463-8.
101. Drets ME, Obe G, Monteverde FJ, Folle GA, Medina II, De-Galvez MG, et al. Computerized graphic and light microscope analyses of T-banded chromosome segments of Chinese hamster ovary cells and human lymphocytes. Biol Zentbl 1992; 111: 204-14.
102. Drets ME, Mendizábal M. The underlying structure of the subtelomeric segments detected by microphotometrical scanning and graphic image analysis. Fundamental and Molecular Mechanisms of Mutagenesis. Mut Res 1998; 404: 13-6.
103. Obe G, Pfeiffer P, Savage JRK, Johannes C, Goedecke W, Jeppesen P, AT, et al. Chromosomal Aberrations: Formation, Identification and Distribution. Mutat Res 2002; 504: 17-36.
104. Drets, ME. Cytological indications on the complex structure of the subtelomeric region. Cytogen Genome Res Chromosomal Aberrations 2004; 104: 137-41.
105. Drets ME, Obe G, Folle GA, Medina II, De-Galvez MG, Duarte, JE, et al. Appearance of "holes" in subtelomeric regions of human and Chinese hamster ovary cell chromosomes due to prolonged incubation in T-banding buffer followed by Giemsa staining. Braz J Genet 1992; 15: 927-33.
106. Drets ME, Mendizábal M, Boccardo EM, Bonomi R. Further analyses of subtelomeric and paracentric holes induced in human and Chinese hamster ovary cell chromosomes. Biol Zentbl 1995; 114: 329-38.
107. Bekaert S, Koll S, Thas O, Van-Oostveldt P. Comparing telomere length of sister chromatids in human lymphocytes using three-dimensional confocal microscopy. Cytometry 2002; 48: 34-44.
108. Badge RM, Yardley J, Jeffreys AJ, Armour JA. Crossover breakpoint mapping identifies a subtelomeric hotspot for male meiotic recombination. Hum Mol Genet 2000; 9: 1239-44.
109. Cornforth MN, Eberle RL. Termini of human chromosomes display elevated rates of mitotic recombination. Mutagenesis 2001; 16: 85-9.
110. Pettenati MJ, Jackle B, Bobby P, Stewart W, Von-Kap-Herr C, Mowrey P, et al. Unexpected retention and concomitant loss of subtelomeric regions in balanced chromosomes anomalies by FISH. Am J Med Genet 2002; 111: 48-53.
111. Mondello C, Pirzio L, Azzalin CM, Giolotto E. Instability of interstitial telomeric sequences in the human genome. Genomics 2000; 68: 111-7.
112. Odero MD, Carlson K, Lahortiga I, Calasanz MJ, Rowley JD. Molecular cytogenetic characterization of breakpoints in 19 patients with hematologic malignancies and 12p unbalanced translocations. Cancer Genet Cytogenet 2003; 142: 115-9.
113. Penas EM, Cools J, Algenstaedt P, Hinz K, Seeger D, Schafhausen P, et al. A novel cryptic translocation t(12;17) (p13;p12-p13) in a secondary acute myeloid leukemia results in a fusion of the ETV6 gene and the antisense strand of the PER1 gene. Genes Chromosomes Cancer 2003; 37: 79-83.
114. Bayani J, Zielenska M, Pandita A, Al-Romaih K, Karaskova J, Harrison K, et al. Spectral karyotyping identifies recurrent complex rearrangements of chromosomes 8, 17, and 20 in osteosarcomas. Genes Chromosomes Cancer 2003; 36: 7-16.
115. Babovic-Vuksanovic D, Jenkins SC, Ensenauer R, Newman DC, Jalal SM. Subtelomeric deletion of 18p in an adult with paranoid schizophrenia and mental retardation. Am J Med Genet 2004; 124: 318-22.
116. Pickard BS, Hollox EJ, Malloy MP, Porteous DJ, Blackwood DH, Armour JA, et al. A 4q35.2 subtelomeric deletion identified in a screen of patients with co-morbid psychiatric illness and mental retardation. BMC Med Genet 2004; 5: 21.
117. Bruyere H, Rajcan-Separovic E, Doyle J, Pantzar T, Langlois S. Familial cryptic translocation (2;17) ascertained through recurrent spontaneous abortions. Am J Med Genet 2003; 123: 285-9.
118. Borg I, Squire M, Menzel C, Stout K, Morgan D, Willatt L, et al. A cryptic deletion of 2q35 including part of the PAX3 gene detected by breakpoint mapping in a child with autism and a de novo 2;8 translocation. J Med Genet 2002; 39: 391-9.
119. Manning MA, Cassidy SB, Clericuzio C, Cherry AM, Schwartz S, Hudgins L, et al. Terminal 2q deletion syndrome: a newly recognized cause of speech and language disability in the autism spectrum. Pediatrics 2004; 114: 451-7.
120. Wolff DJ, Clifton K, Karr C, Charles J. Pilot assessment of the subtelomeric regions of children with autism: detection of a 2q deletion. Genet Med 2002; 4: 10-4.

