










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
Related links
Share

 Permalink
PermalinkRevista Médica del Uruguay
On-line version ISSN 1688-0390
Rev. Méd. Urug. vol.19 no.1 Montevideo May 2003
Importancia de las hipocretinas en la patogenia de la narcolepsia (breve revisión)
Dres. Pablo Torterolo1, Giancarlo Vanini2
Resumen
La hipocretina 1 e hipocretina 2 son neuromoduladores peptídicos que se encuentran en neuronas cuyos somas están localizados en el hipotálamo. Estas neuronas proyectan a diversas regiones del sistema nervioso central. Recientemente se ha descubierto que la alteración de este sistema (sistema hipocretinérgico) se vincula íntimamente con la patogenia de la narcolepsia. Este trabajo pretende hacer una breve reseña de la relación de las hipocretinas con la narcolepsia, así como de la importancia de éstas en la regulación del ciclo sueño-vigilia.
Palabras clave: NARCOLEPSIA.
CATAPLEXIA.
NEUROPÉPTIDOS.
SUEÑO.
VIGILIA.
SUEÑO REM.
HIPOTÁLAMO.
1. Profesor Adjunto de Fisiología.
2. Asistente de Fisiología. Residente de Medicina Intensiva.
Departamento de Fisiología. Facultad de Medicina. Universidad de la República. Montevideo, Uruguay.
Correspondencia: Dr. Pablo Torterolo
Departamento de Fisiología. Facultad de Medicina. Universidad de la República. General Flores 2125. CP 11800, Montevideo, Uruguay.
Email: ptortero@fmed.edu.uy
Recibido: 13/8/02.
Aceptado: 17/1/03.
Introducción
La narcolepsia es una enfermedad neurológica crónica debilitante y hasta ahora incurable, cuya severidad va desde leve a discapacitante(1-8). Por su severidad se la considera una de las principales "enfermedades del sueño". Afecta aproximadamente 1 en 2.000 personas; extrapolando estas cifras existirían 1.500 narcolépticos en Uruguay.
La narcolepsia comienza entre los 15 y 25 años de edad, afectando ambos sexos con similar incidencia. Esta se caracteriza por una completa desorganización del ciclo sueño-vigilia. Los pacientes narcolépticos presentan somnolencia diurna excesiva, cuya forma más habitual es el ataque de sueño. Estos episodios varían de pocos minutos a una hora en su duración y se repiten varias veces en el día. Conjuntamente, es común que los pacientes se sientan adormilados, pasando el día en un incómodo estado de bajo nivel de vigilia. Esto determina escaso rendimiento laboral, lapsos de pérdida de memoria y automatismos motores. Esta hipersomnia constituye el síntoma inicial. Además, estudios polisomnográficos han demostrado que es característico una etapa prematura de sueño REM (siglas en inglés que significan sueño de movimientos oculares rápidos; también llamado sueño paradójico) así como fragmentación del sueño nocturno.
La cataplejia es considerada por algunos autores como el síntoma patognomónico de la narcolepsia. Esta se caracteriza por una atonía muscular súbita provocada por emociones como risa o furia. Puede ser generalizada o parcial de un grupo muscular (caída de la mandíbula o incapacidad de mantener la posición de la cabeza). La conciencia se mantiene durante estos episodios que duran entre 30 segundos y 30 minutos. Sin embargo, clínicamente se distinguen los pacientes narcolépticos con cataplejia (síndrome narcolepsia-cataplejia, 70% de los pacientes narcolépticos) o sin cataplejia. Actualmente se discute si la narcolepsia sin cataplejia constituye una entidad nosológica diferente.
Otros síntomas incluyen parálisis de sueño (incapacidad para moverse durante el inicio del sueño o el despertar) y alucinaciones hipnagógicas (al comienzo del sueño). Estos síntomas aparecen en menos de 30% de los pacientes narcolépticos. A la hipersomnia, la cataplejia, la parálisis del sueño y las alucinaciones hipnagógicas se las considera la "tétrada clínica" de la narcolepsia. Sin embargo, ésta aparece en forma completa en sólo 15% de los pacientes.
Estas manifestaciones producen en el paciente narcoléptico un impacto severo en su calidad de vida en la esfera social (aislamiento) y fundamentalmente laboral (menor rendimiento y accidentes laborales), vinculándose también a mayor probabilidad de accidentes de tránsito.
Recientemente se ha vinculado a las hipocretinas (Hcrt) en la patogenia de la narcolepsia. La importancia de este hallazgo es tal, que se podría afirmar que así como un déficit en el sistema dopaminérgico determina la enfermedad de Parkinson, un déficit en el sistema hipocretinérgico está en la base de la narcolepsia. Esto ha causado un gran entusiasmo en la búsqueda de nuevos métodos diagnósticos y terapéuticos. A su vez, el estudio del sistema hipocretinérgico ha abierto un nuevo campo en la investigación de la fisiología del ciclo sueño-vigilia.
A continuación se hará una breve puesta a punto de la relación del sistema hipocretinérgico con la narcolepsia, así como del papel de este sistema en la regulación del sueño y la vigilia.
Hipocretinas
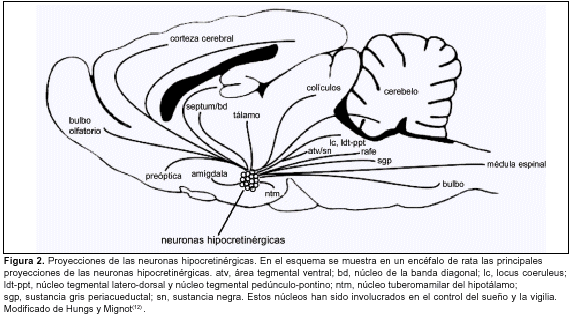
En 1998, dos grupos de investigación independientes identificaron las hipocretinas. La hipocretina 1 y 2, de 33 y 28 aminoácidos respectivamente, son sintetizadas en un pequeño grupo (10.000 a 20.000 neuronas) bilateral y simétrico de neuronas polimorfas localizadas exclusivamente en la región dorsal, posterior y lateral del hipotálamo(9,10) (figura 1). Estas neuronas forman un sistema de proyección difusa que utiliza a las hipocretinas como neuromoduladores y proyecta a diversas regiones del sistema nervioso central (SNC)(11) (figura 2).
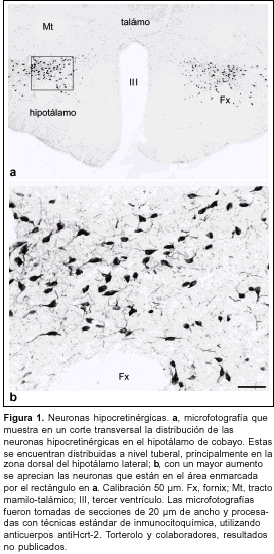
Por su localización en el hipotálamo lateral y su homología con la hormona secretina, se denominó a los péptidos hipocretinas 1 y 2(9). El conocido papel de la región hipotalámica lateral en el control del apetito y el aumento del consumo de alimento luego de la inyección intraventricular de estos neuropéptidos, llevó a otro grupo a denominarlos orexinas A y B (del griego orexis = apetito)(10).
Los dos péptidos son codificados por un único gen precursor de preprohipocretina y ambos péptidos resultan de su clivaje. La Hcrt-1 es idéntica entre ratas, ratones y humanos, mientras la Hcrt-2 humana difiere solo en dos aminoácidos con respecto a la de roedores(12-15).
Existen dos tipos de receptores que son proteínas transmembrana acopladas a proteínas G. El receptor tipo-1 (rHcrt-1) tiene 10 a 100 veces mayor afinidad por la Hcrt-1 que por la Hcrt-2, mientras el receptor tipo-2 (rHcrt-2) une ambas con igual afinidad. Existe una distribución diferente de ambos receptores en distintas regiones del SNC(12-15).
Las hipocretinas producen un efecto excitatorio actuando a través del rHcrt-1 en todos los casos estudiados. Lo mismo sucede actuando sobre el rHcrt-2, aunque se sugiere también que estos podrían actuar como autorreceptores inhibitorios en las neuronas hipocretinérgicas(12-17).
Las hipocretinas en la patogenia de la narcolepsia
Estudios experimentales
Se han obtenido importantes avances en el estudio de la narcolepsia utilizando el modelo de narcolepsia familiar canina. Los perros genéticamente narcolépticos presentan un fenotipo muy similar a la narcolepsia humana(7). Lin y colaboradores(18) observaron que la narcolepsia familiar canina es debida a la mutación del gen del rHcrt-2. Paralelamente, Chemelli y colaboradores(19) realizaron estudios en ratones "knock-out" (en los que se realiza una inactivación o abolición de un gen) para el gen de la preprohipocretina. En estos animales se ven alteraciones comportamentales similares a la narcolepsia; ellos presentan un aumento del tiempo total de sueño, marcada fragmentación del ciclo sueño-vigilia y disminución de la latencia al sueño REM, destacándose, además, frecuentes ataques catapléjicos. Recientemente el síndrome narcoléptico se ha obtenido en ratas lesionando con neurotoxinas las neuronas hipocretinérgicas(20).
Estudios clínicos
Como consecuencia de los descubrimientos en modelos animales se comenzó a estudiar el sistema hipocretinérgico en la narcolepsia humana. El primer estudio fue realizado por Nishino y colaboradores(21) en pacientes caucásicos, observando que los niveles de Hcrt-1 en líquido cefalorraquídeo (LCR) resultaron fácilmente medibles en muestras control, mientras 90% de los pacientes con narcolepsia-cataplejia carecía de Hcrt-1 en el LCR (la Hcrt-2 por su mayor labilidad no se ha logrado medir consistentemente en el LCR). Estos hallazgos fueron confirmados para pacientes japoneses(22). La Hcrt-1 tampoco se detecta al comienzo de la enfermedad en pacientes prepúberes(23); se destaca que normalmente a los 4 meses de edad se encuentran niveles de Hcrt en el LCR similares al de los adultos(24).
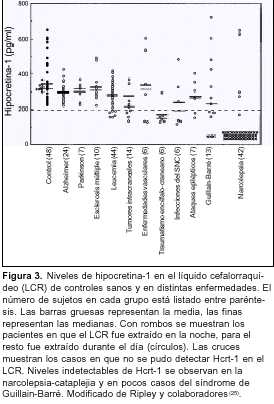
Las enfermedades neurológicas estudiadas presentan niveles de Hcrt-1 en el LCR dentro del rango normal (aproximadamente de 200 a 700 pg/ml)(25) (figura 3). Los niveles de hipocretina también se mantienen normales en la mayoría de los pacientes con narcolepsia sin cataplejia o con otras causas de hipersomnia(26,27). Aunque niveles bajos de Hcrt-1 en el LCR se han observado en ciertos casos de traumatismo encefálico, tumores e infecciones cerebrales, solo en algunos raros casos del síndrome de Guillain-Barré los niveles de Hcrt-1 fueron indetectables(25) (figura 3). Por lo tanto, exceptuando el síndrome de Guillain-Barré, que tiene una presentación clínica diferente a la narcolepsia, se puede decir que la ausencia de Hcrt-1 en el LCR es específico para la narcolepsia-cataplejia(25) (figura 3). Recientemente se ha descrito que los niveles plasmáticos de Hcrt-1 también están disminuidos en la narcolepsia-cataplejia(28).
Estudios de hibridación in situ e inmunohistoquímica en muestras post-mórtem del hipotálamo de pacientes con narcolepsia-cataplejia evidenciaron marcada disminución o ausencia de neuronas hipocretinérgicas. En cambio, solo se encontró una mutación en el gen de la hipocretina en un raro caso narcolepsia-cataplejia de gran severidad, originada a los 6 meses de edad(29). Por lo tanto, aunque existe heterogeneidad en el sector del sistema hipocretinérgico afectado en los distintos casos de narcolepsia-cataplejia, en la mayoría de los casos existe una destrucción de las neuronas hipocretinérgicas, lo que determina una disminución de los niveles de Hcrt-1 en el LCR.
Se piensa que la narcolepsia humana es causada por una compleja interrelación entre factores genéticos y ambientales. En general no existe historia familiar, aunque familiares en primer grado tienen entre 20% y 40% de incremento de riesgo para desarrollar narcolepsia; sólo 25% a 31% de gemelos monocigotos presentan la enfermedad(1). Esta enfermedad se expresa típicamente entre los 12 y 25 años y está estrechamente asociada con el HLA-DR2 y DQB1*0602; la edad de comienzo y la asociación con el complejo mayor de histocompatibilidad sugiere una etiología autoinmune(30). Thannickal y colaboradores(31), estudiando la anatomía patológica de cerebros de pacientes con narcolepsia-cataplejia, describieron un aumento de gliosis en el hipotálamo lateral, consistente con un proceso degenerativo como causa de la pérdida de las neuronas hipocretinérgicas. Este proceso degenerativo es específico para las neuronas hipocretinérgicas ya que no existe degeneración en otras neuronas de la región. Se sostiene que este proceso degenerativo sería de causa autoinmune(32).
Las hipocretinas en el ciclo sueño-vigilia
Se considera que la cataplejia, la parálisis del sueño y las alucinaciones hipnagógicas son resultado de una intrusión de sueño REM, o de algunas de sus manifestaciones, directamente en la vigilia (fisiológicamente el sueño REM aparece luego de una prolongada etapa de sueño lento). Por ejemplo, la cataplejia se considera que se produce por activación durante la vigilia de los sistemas neurales causantes de la atonía característica del sueño REM. Por otra parte, una disminución en la actividad de los sistemas neurales que generan y mantienen la vigilia estaría involucrada en la hipersomnia(1-8).
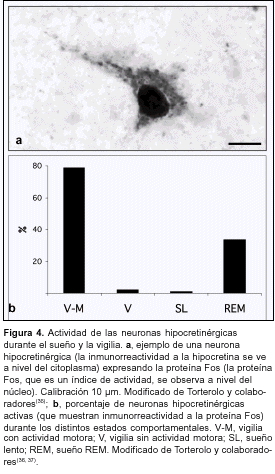
Para comprender la patogenia de la narcolepsia es necesario entender cómo las hipocretinas intervienen normalmente en el control del sueño y la vigilia. Diversas evidencias, entre ellas la importante inervación hipocretinérgica de áreas relacionadas con la generación y el mantenimiento de la vigilia (como el locus coeruleus, el núcleo tuberomamilar del hipotálamo, la región colinérgica mesopontina, etcétera) llevaron a pensar en que este sistema facilitaba la generación y el mantenimiento de la vigilia(12-16). Sin embargo, utilizando la inmunorreactividad a la proteína Fos como índice de actividad neuronal(33), Torterolo y colaboradores han demostrado que las neuronas hipocretinérgicas se activan sólo si existe actividad motora(34-37) (figura 4). Estas neuronas no están activas en vigilia tranquila o en vigilia activada por estímulos sensoriales en ausencia de actividad motora(35,36). Estas neuronas tampoco se encuentran activas durante el sueño lento, un estado en que la actividad motora está disminuida(35,36). Estos resultados han sido confirmados recientemente por Kiyashchenko y colaboradores (2002)(38), quienes utilizando microdiálisis para medir niveles de Hcrt-1 describen un aumento de la liberación de este neuropéptido en varias regiones cerebrales durante la actividad motora. Además, la aplicación intraventricular de hipocretinas aumenta la actividad motora(39). Por lo tanto, el sistema hipocretinérgico no estaría directamente involucrado en la generación y el mantenimiento de la vigilia, sino que su función primaria sería facilitar la actividad motora o el aumento de vigilancia que acompaña a la actividad motora, o ambos. Este aumento de vigilancia estaría mediado por el sistema histaminérgico, ya que la perfusión de hipocretinas en el núcleo tuberomamilar del hipotálamo (donde se encuentran los somas histaminérgicos) aumentan la vigilia(40).
La única enfermedad en que se ha detectado niveles altos de Hcrt-1 en el LCR es el síndrome de piernas inquietas(41), destacándose entonces una posible relación entre el sistema hipocretinérgico y funciones motoras.
Una subpoblación de neuronas hipocretinérgicas se encuentra activa (aumenta su inmunorreactividad a la proteína Fos) durante el sueño REM(34,36) (figura 4). Esto coincide con estudios electrofisiológicos que mostraron un aumento de la frecuencia de descarga de las neuronas de esta región durante el sueño REM(42). Utilizando la técnica de microdiálisis se llegó a la conclusión de que existe un aumento de liberación de Hcrt-1 durante este estado comportamental(38). Además, las neuronas hipocretinérgicas proyectan a la sustancia reticulada pontina medial que es considerada la zona ejecutiva del sueño REM(11); la aplicación de hipocretinas en esta zona facilita su generación(43-45). Resumiendo, las hipocretinas estarían involucradas en la facilitación de la actividad motora y también intervendrían en procesos que ocurren durante el sueño REM. Sin embargo, aunque se ha avanzado en el conocimiento de la fisiología del sistema hipocretinérgico, todavía no se ha aclarado cómo un déficit de este sistema determina el síndrome narcoléptico.
Por último, el sistema hipocretinérgico estaría involucrado en otras funciones, como la regulación de la ingesta(15), el control autonómico(46), el control de la entrada sensorial(47), etcétera.
Perspectivas
El hallazgo de la asociación de narcolepsia con un déficit en el sistema hipocretinérgico abre puertas hacia la búsqueda de recursos diagnósticos y terapéuticos. La cuantificación de niveles de Hcrt-1 en el LCR como método diagnóstico estándar es sensible (aproximadamente 90% de pacientes narcolépticos tiene niveles indetectables de Hcrt-1 en el LCR), específica (solo raros casos del síndrome de Guillain-Barré tienen ausencia de hipocretinas en el LCR) y relativamente económica (los reactivos para radioinmunoanálisis están disponibles comercialmente)(21,25). Este método diagnóstico ya ha encontrado un lugar como complemento de la clínica, junto a la polisomnografía (y al test de múltiples latencias al sueño) y la tipificación de HLA. El reciente hallazgo de que a nivel plasmático la Hcrt-1 está descendida en pacientes narcolépticos-catapléjicos posibilitaría el diagnóstico con un método menos invasivo(28).
En el plano terapéutico existen grandes esperanzas para que la terapia de sustitución o terapia génica, o ambas, sean el futuro tratamiento de esta enfermedad. Con relación a esto, la administración intravenosa de Hcrt-1, que cruza la barrera hematoencefálica(48), produce reversión de síntomas en perros narcolépticos (que tienen una mutación en el receptor rHcrt2)(49). Esta terapia sería más efectiva en pacientes en los que el déficit no está en uno de los receptores sino en la síntesis del neuropéptido. Hasta el momento no existen publicaciones al respecto.
Summary
Hypocretins 1 and 2 are peptidic neuromodulator found in neurons located in the hypothalamus. These neurons carry information to diverse areas of the central nervous system. Recently, it has been discovered that the alteration of the hypocretin system is related to the pathology of narcolepsy. This brief review intends to show the association between hypocretins and narcolepsy, and the role of hypocretins in the regulation of the sleep-wakefulness cycle.
Résumé
Hypocretins 1 and 2 are peptidic neuromodulator found in neurons located in the hypothalamus. These neurons carry information to diverse areas of the central nervous system. Recently, it has been discovered that the alteration of the hypocretin system is related to the pathology of narcolepsy. This brief review intends to show the association between hypocretins and narcolepsy, and the role of hypocretins in the regulation of the sleep-wakefulness cycle.
Bibliografía
1. Aldrich MS. Narcolepsy. N Engl J Med 1990; 323(6): 389-94.
2. Aldrich MS. Narcolepsy. Neurology 1992; 42(7 Suppl 6): 34-43.
3. Aldrich MS. Diagnostic aspects of narcolepsy. Neurology 1998; 50(2 Suppl 1): S2-7.
4. Bassetti C, Aldrich MS. Narcolepsy. Neurol Clin 1996; 14(3): 545-71.
5. Guilleminault C, Anagnos A. Narcolepsy. In: Kryger MH, Roth T, Dement WC (eds.), Principles and practices of sleep medicine. Philadelphia: Saunders, 2000: 676-86.
6. Guilleminault C, Heinzer R, Mignot E, Black J. Investigations into the neurologic basis of narcolepsy. Neurology 1998; 50(2 Suppl 1): S8-15.
7. Mignot E. A hundred years of narcolepsy research. Archives Italiennes de Biologie 2001; 139(3): 207-20.
8. Siegel JM. Narcolepsy: a key role for hypocretins (orexins). Cell 1999; 98: 409-12.
9. de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, et al. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA 1998; 95(1): 322-7.
10. Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 1998; 92: 573-85.
11. Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, et al. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci 1998; 18: 9996-10015.
12. Hungs M, Mignot E. Hypocretin/orexin, sleep and narcolepsy. Bioessays 2001; 23(5): 397-408.
13. de Lecea L, Sutcliffe JG. The hypocretins/orexins: novel hypothalamic neuropeptides involved in different physiological systems. Cell Mol Life Sci 1999; 56(5-6): 473-80.
14. Mignot E. A commentary on the neurobiology of the hypocretin/orexin system. Neuropsychopharmacology 2001; 25(5 Suppl): S5-S13.
15. Willie JT, Chemelli RM, Sinton CM, Yanagisawa M. To eat or to sleep? Orexin in the regulation of feeding and wakefulness. Annu Rev Neurosci 2001; 24: 429-58.
16. Kilduff TS, Peyron C. The hypocretin/orexin ligand-receptor system: implications for sleep and sleep disorders. Trends Neurosci 2000; 23(8): 359-65.
17. Martin G, Fabre V, Siggins GR, de Lecea L. Interaction of the hypocretins with neurotransmitter in the nucleus accumbens. Regul Pept 2002; 104(1-3): 111-7.
18. Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, et al. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell 1999; 98(3): 365-76.
19. Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell 1999; 98(4): 437-51.
20. Gerashchenko D, Kohls MD, Greco M, Waleh NS, Salin-Pascual R, Kilduff TS, et al. Hypocretin-2-saporin lesions of the lateral hypothalamus produce narcoleptic-like sleep behavior in the rat. J Neurosci 2001; 21(18): 7273-83.
21. Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E. Hypocretin (orexin) deficiency in human narcolepsy. Lancet 2000; 355(9197): 39-40.
22. Kanbayashi T, Inoue Y, Chiba S, Aizawa R, Saito Y, Tsukamoto H, et al. CSF hypocretin-1 (orexin-A) concentrations in narcolepsy with and without cataplexy and idiopathic hypersomnia. J Sleep Res 2002; 11(1): 91-3.
23. Tsukamoto H, Ishikawa T, Fujii Y, Fukumizu M, Sugai K, Kanbayashi T. Undetectable Levels of CSF Hypocretin-1 (Orexin-A) in Two Prepubertal Boys with Narcolepsy. Neuropediatrics 2002; 33: 51-2.
24. Kanbayashi T, Yano T, Ishiguro H, Kawanishi K, Chiba S, Aizawa R, et al. Hipocretin-1 (orexin-A)levels in human lumbar CSF in different age groups: infant to elderly persons. Sleep 2002; 25(3): 337-9.
25. Ripley B, Overeem S, Fujiki N, Nevsimalova S, Uchino M, Yesavage J, et al. CSF hypocretin/orexin levels in narcolepsy and other neurological conditions. Neurology 2001; 57: 2253-8.
26. Bassetti CL, Gugger M, Mathis J, Sturzenegger C, Radanov B, Ripley B, et al. Cerebrospinal fluid levels of hypocretin (orexin) in hypersomnolent patients without cataplexy. Actas Fisiol 2001; 7: 205.
27. Overeem S, van Hilten JJ, Ripley B, Mignot E, Nishino S, Lammers GJ. Normal hypocretin-1 levels in Parkinson's disease patients with excessive daytime sleepiness. Neurology 2002; 58: 498-9.
28. Higuchi S, Usui A, Murasaki M, Matsushita S, Nishioka N, Yoshino A, et al. Plasma orexin-A is lower in patients with narcolepsy. Neurosci Lett 2002; 318(2): 61-4.
29. Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med 2000; 6: 991-7.
30. Mignot E, Tafti M, Dement WC, Grumet FC. Narcolepsy and immunity. Adv Neuroimmunol 1995; 5: 23-37.
31. Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, et al. Reduced number of hypocretin neurons in human narcolepsy. Neuron 2000; 27: 469-74.
32. Lin L, Hungs M, Mignot E. Narcolepsy and the HLA region. J Neuroimmunol 2001; 117(1-2): 9-20.
33. Dragunow M, Faull R. The use of c-fos as a metabolic marker in neuronal pathway tracing. J Neurosci Methods 1989; 29(3): 261-5.
34. Torterolo P, Yamuy J, Sampogna S, Morales F, Chase M. C-fos expression in hypocretinergic neurons during wakefulness and carbachol-induced active sleep. Sleep 2001; 24 (Suppl.): A155-6.
35. Torterolo P, Yamuy J, Sampogna S, Morales FR, Chase M. Fos immunoreactivity in hypocretinergic neurons of the cat during active wakefulness, quiet wakefulness and quiet sleep. Actas Fisiol 2001; 7: 196.
36. Torterolo P, Yamuy J, Sampogna S, Morales FR, Chase MH. Hypothalamic neurons that contain hypocretin (orexin) express c-fos during active wakefulness and carbachol-induced active sleep. Sleep Res Online 2001; 4: 25-32. http://www.sro.org/2001/Torterolo/25.
37. Torterolo P, Yamuy J, Sampogna S, Morales FR, Chase MH. Hypocretinergic neurons are primarily involved in activation of the somatomotor system. Sleep 2003; 26(1): 25-8.
38. Kiyashchenko LI, Mileykovskiy BI, Maidment N, Siegel JM. Hypocretin1/Orexin-A release across the sleep-wakefulness cycle. Sleep 2002; 25: 393A.
39. Nakamura T, Uramura K, Nambu T, Yada T, Goto K, Yanagisawa M, et al. Orexin-induced hyperlocomotion and stereotypy are mediated by the dopaminergic system. Brain Res 2000; 873(1): 181-7.
40. Huang ZL, Qu WM, Li WD, Mochizuki T, Eguchi N, Watanabe T, et al. Arousal effect of orexin A depends on activation of the histaminergic system. Proc Natl Acad Sci USA 2001; 98(17): 9965-70.
41. Allen RP, Mignot E, Ripley B, Nishino S, Earley CJ. Increased CSF hypocretin-1 (orexin-A) in restless legs syndrome. Neurology 2002; 59: 639-41.
42. Alam MN, Gong H, Alam T, Jaganath R, McGinty D, Szymusiak R. Sleep-waking discharge patterns of neurons recorded in the rat perifornical lateral hypothalamic area. J Physiol 2002; 538(Pt 2): 619-31.
43. Bourgin P, Huitron-Resendiz S, Spier AD, Fabre V, Morte B, Criado JR, et al. Hypocretin-1 modulates rapid eye movement sleep through activation of locus coeruleus neurons. J Neurosci 2000; 20(20): 7760-5.
44. Xi MC, Morales FR, Chase MH. Effects on sleep and wakefulness of the injection of hypocretin-1 (orexin-A) into the laterodorsal tegmental nucleus of the cat. Brain Res 2001; 901: 259-64.
45. Yamuy J, Xi MC, Fung SJ, Torterolo P, Zhang J, Rojas MJ, et al. Effects of hypocretin (orexin) in pontine nuclei involved in active sleep. Actas Fisiol 2001; 7: 57.
46. Lin Y, Matsumura K, Tsuchihashi T, Abe I, Iida M. Chronic central infusion of orexin-A increases arterial pressure in rats. Brain Res Bull 2002; 57(5): 619-22.
47. Vanini G, Torterolo P, Falconi A, Velluti RA. The hypothalamic hypocretinergic system modulates auditory unit firing at the inferior colliculus. Sleep 2003 (en prensa).
48. Kastin AJ, Akerstrom V. Orexin A but not orexin B rapidly enters brain from blood by simple diffusion. J Pharmacol Exp Ther 1999; 289(1): 219-23.
49. John J, Wu M, Siegel JM. Systemic administration of hipocretin-1 reduces cataplexy and normalizes sleep and waking durations in narcoleptic dogs. Sleep Res Online 2000; 3(1): 23-8.

