











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Introducción.
La fenilcetonuria (PKU) es un error innato del metabolismo de transmisión autosómica recesiva ocasionado por variantes patogénicas en el gen PAH, que codifica la enzima hepática fenilalanina hidroxilasa (PAH). La pérdida parcial o total de la actividad enzimática impide la hidroxilación de fenilalanina (Phe) a tirosina (Tyr), lo que conlleva a su acumulación neurotóxica en suero y tejidos, particularmente en el sistema nervioso central1. Esta variabilidad da lugar a un amplio espectro bioquímico, que abarca desde la hiperfenilalaninemia benigna y moderada hasta la PKU clásica. Aunque el neonato es asintomático al nacimiento, la exposición sostenida a niveles elevados de Phe provoca daño neurológico irreversible si no se instaura tratamiento dietético oportuno2.
El diagnóstico temprano se realiza mediante la determinación de los niveles de fenilalanina en sangre seca recolectada entre las 24 y 72 horas de vida. Desde 2011, el programa nacional de tamizaje metabólico del Ecuador permite la identificación precoz de esta entidad y la prevención de sus complicaciones3. Sin embargo, el éxito terapéutico depende del diagnóstico precoz, del suministro oportuno de fórmulas medicadas, del seguimiento multidisciplinario y de la educación continua a la familia2.
En ausencia de tratamiento, los pacientes con PKU pueden desarrollar discapacidad intelectual grave, microcefalia adquirida, convulsiones, alteraciones conductuales, hipopigmentación difusa y un característico olor corporal a humedad o moho4. A continuación, se describe el caso de una lactante de seis meses con PKU clásica, destacando las barreras de acceso al tratamiento y su evolución clínica y bioquímica tras la instauración de una terapia nutricional especializada.
Presentación del caso clínico
Se trata de una lactante femenina de siete meses de edad, procedente de Santo Domingo de los Tsáchilas (Ecuador), producto de la segunda gesta de una madre con controles prenatales adecuados. Nació por parto eutócico a las 39 semanas, con peso 3 540 g, talla 50 cm y perímetro cefálico 35 cm; Apgar 8/9. Sin antecedentes familiares relevantes ni consanguinidad parental.
El tamizaje metabólico neonatal se realizó a los 7 días de vida, reportando niveles de fenilalanina de 841 µmol/L, valor sospechoso para PKU. Una segunda medición por cromatografía líquida a los 44 días reveló 1248 µmol/L de fenilalanina y 60 µmol/L de tirosina, compatible con hiperfenilalaninemia severa. El examen físico y neurológico fue normal.
Tras la confirmación de fenilcetonuria clásica se gestionó referencia a un centro especializado; sin embargo, la demora en la provisión de la fórmula libre de Phe prolongó la lactancia materna exclusiva durante seis semanas adicionales, sin intervención dietética específica, lo cual favoreció la persistencia de niveles plasmáticos elevados con niveles de fenilalanina de 1248 µmol/L a las 11 semanas 4 días 1287 µmol/L. La suplementación con fórmula metabólica (5 g/60 mL, 5 tomas/día durante 15 días; luego 10 g/180 mL cada 4 h) inició a la semana 14 semanas de vida, con descenso de fenilalanina a 1 082,5 µmol/L y posteriormente a 731,5 µmol/L a la semana 16 de vida, descritos en elgráfico 1.
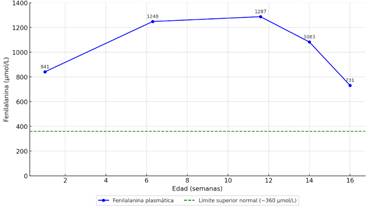
Gráfico 1: Evolución de los niveles plasmáticos de fenilalanina. Seguimiento de los niveles de fenilalanina (µmol/L) desde la primera hasta la semana 16 de vida. Se observa un aumento sostenido en ausencia de tratamiento, seguido de un descenso tras iniciar fórmula metabólica especializada. La línea verde discontinua marca el límite superior normal (~360 µmol/L), superado en todo momento durante el seguimiento.
A los seis meses se inició la alimentación complementaria supervisada, manteniendo fórmula libre de fenilalanina a libre demanda y suplementación con vitaminas A, C y D, con introducción progresiva de alimentos naturales bajos en proteínas. Los exámenes bioquímicos se mantuvieron dentro de los parámetros normales, descritos en la tabla 1.
Tabla 1: Exámenes complementarios
| Parámetro | Resultado | Unidad | Valores de Referencia | Parámetro | Resultado | Unidad | Valores de Referencia |
| SERIE BLANCA | SERIE ROJA | ||||||
| Leucocitos | 7.50 | 10³/µl | 6.00 - 17.50 | Hematíes | 4.56 | 10⁶/µl | 3.20 - 5.20 |
| Neutrófilos % | 22.3 | % | 17.0 - 60.0 | Hemoglobina | 11.0 | g/dl | 10.1 - 12.9 |
| Linfocitos % | 72.1 | % | 20.0 - 70.0 | Hematocrito | 35.8 | % | 32.0 - 44.0 |
| Eosinófilos % | 2.8 | % | 1.0 - 5.0 | VCM | 78.4 | Fl | 73.0 - 109.0 |
| Monocitos % | 2.4 | % | 1.0 - 11.0 | HCM | 24.10 | Pg | 21.0 - 33.0 |
| Basófilos % | 0.4 | % | 0.0 - 1.0 | CHCM | 30.7 | g/dl | 26.0 - 39.0 |
| Total, de Morfología | 100.0 | % | - | Plaquetas | 393 | 10³/µl | 229 - 553 |
| BIOQUIMICA SANGUINEA | |||||||
| Glucosa | 98.4 | mg/dl | 60 - 110 | TGO/AST | 45.5 | U/L | Hasta 77.0 |
| Urea | 15.1 | mg/dl | 8.6 - 40.7 | TGP/ALT | 40.1 | U/L | Hasta 56.0 |
| BUN | 7.1 | mg/dl | 10 - 20 | Colesterol total | 127.9 | mg/dl | Hasta 200 |
| Creatinina | 0.21 | mg/dl | 0.17 - 0.42 | Triglicéridos | 46.2 | mg/dl | Hasta 150 |
| Ácido úrico | 2.35 | mg/dl | 3.00 - 6.10 | Proteínas Totales | 6.45 | g/dl | 4.80 - 7.60 |
Durante el seguimiento, la paciente mostró un desarrollo neuromadurativo acorde a su edad cronológica, sin signos de retraso global del desarrollo. Esto fue corroborado mediante evaluación clínica especializada y herramientas psicométricas validadas, como Denver II y la Escala Bayley-III, evidenciando puntajes dentro del rango normativo en dominios cognitivo, motor (grueso y fino), y del lenguaje (receptivo y expresivo) descritos en el Gráfico 2.
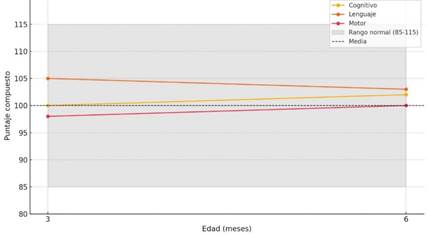
Gráfico 2: Seguimiento del neurodesarrollo a los 3 y 6 meses utilizando los puntajes compuestos de la Escala Bayley-III. Se observa que todos los dominios evaluados se mantienen dentro del rango normal (85-115), lo que indica un desarrollo adecuado hasta el momento.
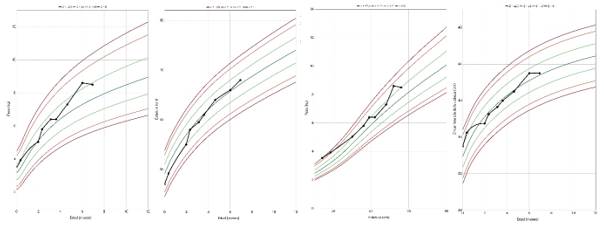
El crecimiento somático se mantuvo dentro de los percentiles esperados según las curvas de crecimiento de la OMS (Z-score 0 a +1), descrito en el gráfico 3. Se aplicó el cuestionario PKU-QOL en su versión para cuidadores, observándose una adecuada adherencia al tratamiento hipoproteico, con baja carga emocional familiar. Sin embargo, persistió la preocupación parental por la falta de normalización de los niveles plasmáticos de fenilalanina. La paciente continúa bajo vigilancia multidisciplinaria por endocrinología, nutrición clínica, genética médica y neurología pediátrica, con reevaluaciones periódicas del desarrollo y de la calidad de vida.
Discusión
Latinoamérica está conformado por 33 países, de los cuales solo 20 disponen de programas de tamizaje neonatal. Sin embargo, solo paises como Cuba, Costa Rica, Chile y Uruguay alcanzan una cobertura superior al 99 %, mientras que Honduras y República Dominicana la cobertura no supera el 5%. Está disparidad se refleja en las tasas de mortalidad infantil, que oscilan entre 3.7 y 6.4 por mil nacidos vivos en los países con alta cobertura, frente a tasas de 8.8 a 49,5 por mil en aquellos con menor alcance5.
En este contexto, Ecuador se ha consolidado como un referente regional en la implementación sostenida del tamizaje metabólico neonatal. Desde el inicio del programa nacional en 2011, se han tamizado más de 2,6 millones de neonatos, alcanzando una cobertura del 82,5 % hasta octubre del 2024. Actualmente, el protocolo nacional incluye la detección precoz de cuatro errores innatos del metabolismo: hipotiroidismo congénito, hiperplasia suprarrenal congénita, galactosemia y fenilcetonuria. Estas patologías, han sido diagnosticadas e intervenidas oportunamente en más de 730 lactantes, permitiendo iniciar tratamiento antes de los 30 días de vida5,6.
La fenilcetonuria (PKU) es una enfermedad de herencia autosómica recesiva, causado por mutaciones en el gen PAH (12q22-q24.2), que codifica para la enzima hepática fenilalanina hidroxilasa. Esta enzima cataliza la conversión de fenilalanina (Phe) en tirosina (Tyr). La deficiencia o disfunción de enzima fenilalanina hidroxilasa (PAH) produce acumulación sistémica de fenilalanina7. Este caso resalta la importancia del tamizaje neonatal, que permitió identificar una hiperfenilalaninemia severa en una recién nacida asintomática. No obstante, se evidenciaron barreras estructurales en el acceso al tratamiento, lo que retrasó el inicio de la terapia nutricional especializada, exponiendo a la paciente a niveles neurotóxicos de fenilalanina durante un período crítico del desarrollo cerebral.
Aunque se considera una enfermedad rara, la PKU es uno de los trastornos metabólicos hereditarios más frecuentes, con una incidencia global estimada de 1 por cada 23 930 nacidos vivos y afecta a aproximadamente 0,45 millones de personas7. No obstante, se observan marcadas diferencias étnicas, la tasa más alta se registran en poblaciones europeas, particularmente en Irlanda (1 en 4 500), mientras que la tasa más baja se observa en Finlandia (1 en 200 000). La prevalencia en América del Sur varía de 1:25.000 a 50.000 nacidos vivos, con una prevalencia menor en la parte norte que en la parte sur del continente1,7. En este contexto, en países como Ecuador, se han identificado 55 casos confirmados de fenilcetonuria desde el inicio del Programa Nacional de Tamizaje Metabólico en el año 2011 hasta octubre de 2024. Esta cifra corresponde a una prevalencia estimada de 1,2 en 10.000 nacidos vivos3,6 .
Un estudio realizado en México y publicado en 2021 evaluó a 124 pacientes diagnosticados con fenilcetonuria, de estos, el 78.2% fue identificado de manera temprana gracias al tamizaje neonatal, mientras que el 21.8% fue diagnosticado a través de un enfoque clínico. En cuanto a los niveles de fenilalanina en sangre, se determinó que el 50% de los pacientes presentaba fenilcetonuria clásica (> 1200 µmol/L), el 20.2% tenía una forma moderada de PKU (360-1140 µmol/L), y el 29.8% mostraba hiperfenilalaninemia leve (240-360 µmol/L) (9. En referencia a este estudio, el caso clínico se enmarca en una fenilcetonuria clásica, con concentraciones plasmáticas de fenilalanina de 1248 µmol /l y tirosina de 60 µmol /l a los 44 días de vida.
Al momento del diagnóstico, es esencial restringir la ingesta de proteínas, especialmente aquellas ricas en fenilalanina, mediante el uso de fórmulas libres de melanina y alimentos que aporten menos de 75 mg de fenilalanina por cada 100 g, descritos en la tabla 2 10,11. En este contexto, la leche materna contiene una concentración relativamente baja de fenilalanina (aproximadamente 46 mg por cada 100 ml), representa una opción nutricional adecuada durante los primeros seis meses de vida. Su uso cobra especial relevancia en situaciones donde no se dispone de fórmulas especiales libres de fenilalanina, ya sea por un diagnóstico tardío o por barreras socioeconómicas que limitan el acceso8,12. Sin embargo, debido a la variabilidad en los niveles de fenilalanina en la leche materna y la dificultad de medir con precisión el volumen de leche consumido por el bebé, se requiere una supervisión cuidadosa y constante de los niveles de fenilalanina en la sangre13.
Tabla 2: Alimentos bajos en proteínas que se pueden comer sin restricciones en una dieta baja en fenilalanina
| Grupo de alimentos | Alimentos permitidos sin restricción | Límite de proteínas permitido |
| Frutas y verduras | Todas las frutas y verduras con ≤ 75 mg de fenilalanina/100 g (excepto papas) | - |
| Grasas | Mantequilla, margarina, ghee, aceites vegetales y productos grasos con ≤ 1 g proteína/100 g | ≤ 1 g proteína/100 g |
| Almidones | Harina de yuca, arrurruz, sagú, tapioca, almidón de maíz con ≤ 0.5 g proteína/100 g | ≤ 0.5 g proteína/100 g |
| Queso vegano | Queso vegano con ≤ 0.5 g proteína/100 g | ≤ 0.5 g proteína/100 g |
| Azúcares | Azúcar, glucosa, miel, mermelada, jarabes, sorbetes, paletas sin proteína significativa | ≤ 0.5 g proteína/100 g |
| Gelatina veg/agar sin gelatina | Gelatina/agar con ≤ 0.5 g proteína/100 g | ≤ 0.5 g proteína/100 g |
| Alimentos especiales bajos en proteínas | Harinas, mezclas, galletas, pastas y otros si todos los ingredientes son bajos en proteínas y fenilalanina | ≤ 25 mg fenilalanina/100 g |
| Hierbas/especias | Todas, ya que se usan en cantidades muy pequeñas | - |
| Bebidas | Refrescos, jugos, tés, agua, etc., siempre que no contengan aspartamo | - |
| Leche especial baja en proteínas y en Phe | Leches con ≤ 25 mg fenilalanina en 24 h del volumen consumido | ≤ 25 mg fenilalanina/24 h |
| Leche vegetal | Leches vegetales con ≤ 0.1 g proteína/100 g (por ejemplo, almendra) | ≤ 0.1 g proteína/100 g |
| Misceláneas | Esencias alimentarias y colorantes usados en pequeñas cantidades | - |
Adaptado de: MacDonald A, et al. Fenilcetonuria dietary handbook to accompany PKU guidelines. Orphanet J Rare Dis. 2020.
No existe un consenso definitivo sobre el manejo alimentario en recién nacidos y lactantes con PKU, las guías europeas recomiendan fomentar la lactancia materna combinadas con fórmulas libres de fenilalanina, mientras que las directrices del Reino Unido no brindan orientaciones claras al respecto12,14. En la práctica clínica, se han descrito dos modalidades para ofrecer los beneficios de la lactancia: una consiste en administrar primero la formula metabólica especial y luego permitir la lactancia materna a libre demanda con un máximo de 8 tomas diarias; la otra, se centra en administrar leche materna extraída en cantidades controladas junto con la fórmula, pero esta última opción pierde el estímulo directo del seno materno15. Según la Guía de Práctica Clínica del Ministerio de Salud Pública de Ecuador, todos los niños diagnosticados con PKU deben recibir fórmulas especiales libres de fenilalanina, proporcionadas en los establecimientos de atención primaria. La indicación, tipo y cantidad del sustituto dependen de la clasificación de la enfermedad, la edad y el estado nutricional del paciente descritos en latabla 3 16.
Tabla 3: Requerimientos de Sustituto Lácteo en Pacientes Pediátricos con PKU
| Grupo de edad | Peso estimado | Requerimiento proteico total | Proteínas del sustituto (70%) | Cantidad de polvo diario |
| 0 a 17 meses 29 días | 3 - 10 kg | 2,5 g/kg/día | 5 - 14 g/día | 40 - 90 g/día |
| 18 meses a 5 años 11 meses 29 días | 13 - 20 kg | 16 - 25 g/día | 14 - 21 g/día | 60 - 80 g/día |
| 6 a 7 años 11 meses 29 días | 20 - 28 kg | >30 g/día (≈1,8 g/kg/día) | 21 - 25 g/día | 90 g/día |
| 8 a 9 años 11 meses 29 días | 25 - 35 kg | ≈60 g/día | 42 g/día | 100 g/día |
| 10 a 13 años 11 meses 29 días | 45 - 50 kg | 72 g/día | 42 - 50 g/día | 100 - 120 g/día |
Adaptado de: Ministerio de Salud Pública del Ecuador. Guía de práctica clínica: Diagnóstico y tratamiento nutricional del paciente pediátrico y adolescente con PKU. Quito - Ecuador; 2013.
En el caso clínico descrito, se implementó un esquema terapéutico combinado que incluyó lactancia materna a libre demanda, complementada con una fórmula metabólicamente especializada, libre de fenilalanina y formulada con una mezcla de aminoácidos esenciales y no esenciales, carbohidratos, lípidos, vitaminas, minerales y oligoelementos, destinada al soporte nutricional en pacientes con fenilcetonuria.
El protocolo de administración se inició con 5 g del producto disueltos en 60 ml de agua, cinco veces al día durante 15 días, posteriormente ajustado a 10 g en 180 ml cada 4 horas, conforme a los requerimientos proteicos individuales y la tolerancia metabólica de la paciente. Dicho esquema permitió una disminución de los niveles plasmáticos de fenilalanina a 731,48 µmol/L a las 16 semanas de vida. Sin embargo, según las guías europeas actuales, el tratamiento de la PKU debe mantenerse de por vida, con el objetivo de mantener las concentraciones plasmáticas de fenilalanina entre 120-360 µmol/L en niños hasta los 12 años, y entre 120-600 µmol/L en adolescentes y adultos. Estas directrices tienen como finalidad prevenir alteraciones del desarrollo neurológico y proteger funciones críticas como la mielinización y la síntesis de neurotransmisores, particularmente dopamina y serotonina, fundamentales para el funcionamiento cerebral17.
En respuesta a estas limitaciones, se han desarrollado terapias farmacológicas como el diclorhidrato de sapropterina y la pegvaliasa. La sapropterina, indicada en pacientes con respuesta a tetrahidrobiopterina, mejora la actividad de la fenilalanina hidroxilasa residual y permite una mayor ingesta de proteínas naturales17,18. No obstante, su uso varía entre países debido a la ausencia de protocolos estandarizados. Por su parte, la pegvaliasa, una enzima recombinante pegilada, convierte la fenilalanina en compuestos no tóxicos de forma independiente de la actividad enzimática endógena19. Sin embargo, ambas terapias enfrentan limitaciones en países en desarrollo, principalmente por sus altos costos, la necesidad de seguimiento especializado y la limitada disponibilidad.
El control metabólico se asocia con el desempeño neurocognitivo a largo plazo. Estudios recientes subrayan la necesidad de evaluaciones periódicas del coeficiente intelectual en la adolescencia y un seguimiento neuropsiquiátrico individualizado, considerando factores como el rendimiento académico, el estado emocional y la adherencia a la dieta17. La evaluación de funciones ejecutivas, memoria y atención resulta esencial, ya que las alteraciones en estas áreas son comunes en pacientes con PKU no controlada. Sin embargo, aunque la resonancia magnética cerebral puede mostrar alteraciones, no se ha establecido una correlación directa entre los hallazgos radiológicos y el control de los niveles de fenilalanina. Esto sugiere que dicho examen debe reservarse para casos con manifestaciones neurológicas claras19.
La fenilcetonuria en ausencia de un diagnóstico y tratamiento oportuno conlleva consecuencias neurológicas severas e irreversibles. En los primeros meses de vida, los lactantes suelen permanecer asintomáticos, sin embargo, a partir del segundo semestre, la acumulación progresiva de fenilalanina en el sistema nervioso central ocasiona neurotoxicidad, con daño estructural y funcional. Este deterioro neurológico se manifiesta a través de retraso en el desarrollo psicomotor, compromiso del lenguaje, disminución del coeficiente intelectual y trastornos en la atención y el control motor. Si bien el control metabólico mediante una dieta restringida en fenilalanina constituye el pilar del tratamiento, la exigencia de adherencia prolongada y estricta impacta negativamente en la calidad de vida del paciente. Además, factores psicosociales, como el estigma asociado a una enfermedad crónica y las limitaciones dietéticas impuestas desde la infancia, agravan esta carga20.
El inicio tardío del tratamiento dietético en la PKU representa un factor de riesgo significativo para el desarrollo de secuelas neurológicas sutiles pero persistentes. En este contexto, el seguimiento neuropsicológico continuo se vuelve esencial, especialmente en pacientes con diagnóstico demorado o manejo dietético irregular. Diversos estudios han evidenciado que dichas alteraciones pueden manifestarse incluso en pacientes bajo tratamiento, afectando funciones ejecutivas como la memoria de trabajo y el control inhibitorio desde etapas tempranas del desarrollo. Paermentier et al. (2023) identificaron estas deficiencias en una cohorte de niños en edad preescolar con PKU, destacando la presencia de alteraciones neurocognitivas que podrían pasar desapercibidas sin una evaluación especializada21. De manera similar, un estudio retrospectivo en una cohorte portuguesa revelo que un control metabólico subóptimo -con niveles de fenilalanina superiores a 7.8 mg/dL a los tres años- predice un menor rendimiento cognitivo a los seis años22.
Estos hallazgos subrayan la importancia de implementar periódicamente herramientas neuropsicológicas estandarizadas, como la Escala de Desarrollo Mental de Griffiths o el BRIEF-P, desde el diagnóstico y a lo largo del crecimiento. Esta estrategia permite la detección temprana de déficits emergentes e impulsa intervenciones personalizadas que optimizan el pronóstico funcional y favorecen la inclusión social, particularmente en adolescentes y adultos jóvenes con historial de manejo tardío o intermitente20.
Conclusión
La PKU representa un trastorno metabólico grave pero prevenible, cuya detección mediante programas de tamizaje neonatal es esencial para un diagnóstico oportuno. Este caso clínico refuerza el valor del diagnóstico temprano y del inicio inmediato de una dieta restringida en fenilalanina para prevenir daños neurológicos irreversibles. Como nuevo conocimiento se destaca la viabilidad del uso combinado de la lactancia materna y formulas especiales en el manejo nutricional, así como la respuesta favorable al tratamiento en etapas iniciales. Estos hallazgos se alinean con las recomendaciones de las guías europeas actuales, que promueven la lactancia materna a libre demanda combinada con fórmulas especiales sin fenilalanina.
Una de las limitaciones de este estudio es que, al ser un único caso clínico, restringe la generalización de sus conclusiones. Además, quedan pendientes aspectos como la estandarización de protocolos de alimentación en lactantes con PKU y el acceso garantizado a suplementos metabólicos, aun ausenten en el cuadro básico de medicamentos del Ecuador. Por tanto, son necesarias investigaciones adicionales, tanto clínica como de políticas públicas, que evalúen estrategias de manejo nutricional y aseguren la disponibilidad de los suplementos alimenticios para mejorar la calidad de vida de estos pacientes desde los primeros días de vida.

