











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Introducción
La atrofia muscular espinal (AME) es un grupo de trastornos neuromusculares hereditarios caracterizados por degeneración de las neuronas motoras alfa del asta anterior de la médula espinal, lo que conlleva a atrofia muscular progresiva, debilidad y parálisis1.
La forma más común, la AME tipo 5q, se debe a una alteración en el gen SMN1, ubicado en 5q11.2-q13.32. En aproximadamente el 95% de los casos esta alteración corresponde a una deleción en homocigosis del exón 7 del gen; en el restante 5% se detectan mutaciones puntuales. Por esta localización, se conoce como AME 5q3,4. El gen SMN1 codifica la proteína de supervivencia de la motoneurona (SMN), cuya ausencia o déficit conlleva a la apoptosis acelerada de las motoneuronas. Existe un gen homólogo, SMN2, que por un cambio en el sitio de splicing del exón 7 produce mayoritariamente una proteína truncada y no funcional. Sin embargo, entre un 10%-15% del ARNm de SMN2 incluye el exón 7, permitiendo la síntesis parcial de proteína funcional4-6. Existe cierta correlación genotipo-fenotipo: menor número de copias de SMN2 se asocia con formas más graves de la enfermedad3,4,7,8.
El diagnóstico se realiza mediante estudio genético. Ante la sospecha clínica, se recomienda inicialmente la técnica MLPA para detectar deleción del exón 7 en SMN1; si es negativa, debe completarse con secuenciación completa del gen para descartar mutaciones puntuales8-11.
Hasta el desarrollo de terapias modificadoras de la enfermedad, el tratamiento de la AME era puramente sintomático. El primer tratamiento específico aprobado fue nusinersen, autorizado por la Food and Drug Administration (FDA) en 2016 y por la Agencia Europea de Medicamentos (EMA) en 20179-14. En Uruguay, su uso comenzó en enero de 2019, inicialmente mediante recursos judiciales. En mayo de 2022 fue incorporado formalmente a través del Fondo Nacional de Recursos (FNR). La administración de nusinersen puede realizarse por punción lumbar convencional o, en casos con dificultad anatómica o posquirúrgica, a través de un reservorio intratecal adaptado (como el reservorio de Ommaya conectado al espacio subaracnoideo). La posología recomendada incluye cuatro dosis de carga (días 0, 14, 28 y 63), seguidas de una dosis de mantenimiento cada cuatro meses15.
En el seguimiento clínico se utilizan escalas funcionales validadas, las cuales permiten cuantificar la progresión motora en relación con la historia natural de la enfermedad. En AME tipo 1 se utilizan CHOP INTEND (rango 0-64 puntos) y HINE-2 (hasta 26 puntos), mientras que en AME tipo 2 se emplean HFSME (hasta 66 puntos) y RULM (hasta 37 puntos), donde mayores valores indican mejor desempeño motor.
El objetivo de este estudio es describir la evolución clínica y las características del primer grupo de pacientes en Uruguay que recibió tratamiento con nusinersen, marcando así el inicio de una nueva etapa terapéutica en el abordaje de la AME en el país.
Metodología
Se realizó un estudio descriptivo, observacional y prospectivo. La población de estudio estuvo constituida por el primer grupo de pacientes con AME evaluados en distintos servicios asistenciales del Sistema Nacional Integrado de Salud (SNIS) de Uruguay, que iniciaron tratamiento con nusinersen indicado por su neuropediatra tratante.
Los criterios de inclusión fueron:
• Pacientes con alteración del gen SMN1 comprobada por estudio genético, diagnosticados con AME tipo 1 o 2 en el período comprendido entre enero de 2019 y junio de 2022 inclusive.
• Acceso completo a datos clínicos antes y después del inicio del tratamiento.
• Haber recibido al menos un año de tratamiento con nusinersen y haber cumplido con el protocolo de evaluación funcional: al inicio, a los seis meses y al año. Si se disponía de evaluación a los dos años, también fue incluida.
• Seguimiento periódico y regular por equipos interdisciplinarios del SNIS.
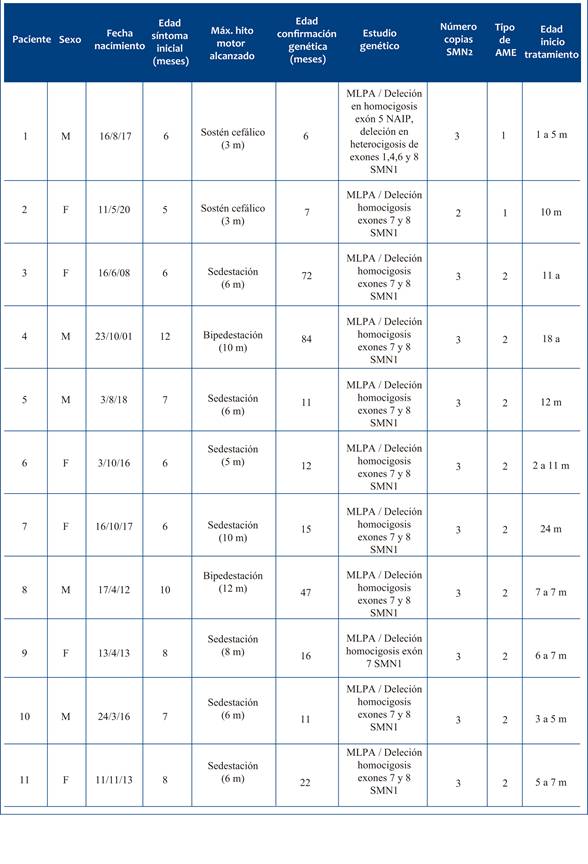
Se excluyeron los pacientes que no cumplieran el tiempo mínimo de tratamiento, no concurrieran a controles posteriores o no hubieran otorgado consentimiento/asentimiento informado. Se relevaron las siguientes variables: sexo, edad, edad de inicio de síntomas, síntoma inicial, edad al diagnóstico genético, tipo de estudio realizado, resultado genético, número de copias del gen SMN2, tipo de AME, máximo hito motor alcanzado (sostén cefálico, sedestación, bipedestación, deambulación), edad y fecha de inicio de tratamiento, número total de dosis de nusinersen, efectos adversos, tipo de soporte nutricional (vía oral, sonda nasogástrica, gastrostomía), soporte respiratorio (espontáneo, fisioterapia, asistencia respiratoria), complicaciones ortopédicas (escoliosis, cirugía, fisioterapia) y escalas de valoración funcional motora (CHOP INTEND, HINE-2, HFSME, RULM).
Los datos se recolectaron mediante revisión de historia clínica y se analizaron mediante planillas electrónicas. Las variables cuantitativas son representadas con medidas de tendencia central y su dispersión (media con su desvío estándar), y las variables cualitativas se expresan en frecuencias absolutas y relativas. Se exploraron asociaciones entre variables mediante razón de probabilidades (odds ratio, OR) y riesgo relativo (RR) con su intervalo de confianza de 95%.
Debido al tamaño reducido de la muestra, los análisis estadísticos realizados tienen un carácter exploratorio y descriptivo, sin intención inferencial. La comparación de medias entre grupos se realizó mediante test de t de Student, considerando un valor de p<0,05 como indicativo de diferencia potencialmente significativa.
Resultados
Entre enero de 2019 y junio de 2022, 15 pacientes iniciaron tratamiento con nusinersen mediante recurso de amparo judicial, ya que en ese momento el fármaco no estaba incluido en el marco de prestaciones del SNIS. Se presentan aquí los resultados de 11 pacientes que cumplieron con los criterios de inclusión previamente establecidos. Los casos restantes fueron excluidos según dichos criterios.
Del total incluido, 6 (54,5%) eran de sexo femenino. La edad de aparición del primer síntoma presentó una mediana de 7 meses, con un rango de 6 a 12 meses. En todos los casos la presentación clínica inicial fue debilidad muscular. Todos los pacientes fueron sometidos a estudio genético mediante técnica MLPA. Nueve (81,8%) presentaron deleción en homocigosis de los exones 7 y 8 del gen SMN1. Los dos restantes presentaron combinaciones atípicas: uno con deleción en homocigosis del exón 5 del gen NAIP y en heterocigosis de los exones 1, 4, 6 y 8 del SMN1; el otro, deleción en homocigosis del exón 7 del SMN1. Diez pacientes (90,9%) tenían tres copias del gen SMN2, y uno tenía dos copias. La edad de confirmación genética osciló entre los 6 meses y los 7 años.
Según la clasificación clínica, 2 pacientes (18,2%) fueron diagnosticados con AME tipo 1, y 9 (81,8%) con AME tipo 2. El hito motor máximo alcanzado en los pacientes con AME tipo 1 fue el sostén cefálico; en los pacientes con AME tipo 2, se alcanzó la bipedestación.
En cuanto al inicio del tratamiento, los dos pacientes con AME tipo 1 comenzaron la terapia a los 10 y 17 meses, respectivamente. En los pacientes con AME tipo 2, el inicio ocurrió entre 1 y 18 años. En algunos casos el tratamiento se demoró por razones administrativas o sanitarias. El paciente 1 fue el primero en recibir nusinersen en Uruguay, antes de su aprobación oficial, lo cual explica el retraso. En el caso del paciente 2, la pandemia por COVID-19 impactó en los tiempos de diagnóstico e inicio de tratamiento. Algunos de los pacientes, conocidos como “históricos”, habían sido diagnosticados años antes de la aprobación y comenzaron tratamiento una vez que el medicamento estuvo disponible a nivel nacional con el objetivo de evaluar la respuesta clínica (Tabla 1).
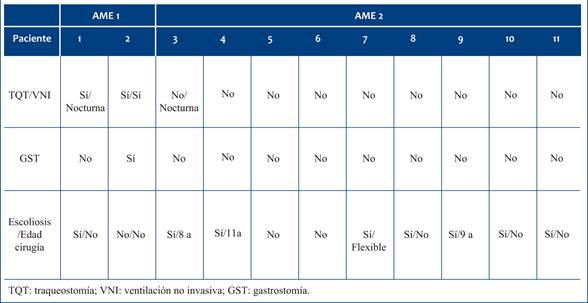
La mayoría de los pacientes recibió nusinersen por vía habitual. Dos de ellos, debido a cirugía de columna, requirieron la colocación de un reservorio Ommaya para la infusión intratecal. Los efectos adversos reportados fueron leves: 4 pacientes (36,4%) presentaron cefalea pospunción y 1 (9%) mostró elevación transitoria de aminotransferasas sin otra causa identificable. Ningún paciente tratado por reservorio presentó efectos secundarios.
En la (Tabla 2) se resumen las complicaciones observadas. En el grupo AME tipo 1, ambos pacientes iniciaron tratamiento con traqueotomía (TQT) y ventilación mecánica permanente; uno de ellos logró suspender la ventilación diurna. Ningún paciente con AME tipo 2 requirió TQT y solo uno usa ventilación nocturna. En cuanto al soporte nutricional, uno de los pacientes con AME tipo 1 recibió gastrostomía (GST). Ningún paciente tipo 2 la requirió. Con respecto a complicaciones ortopédicas, un paciente AME tipo 1, y siete pacientes AME tipo 2 (72,7%) desarrollaron escoliosis, de los cuales tres fueron intervenidos quirúrgicamente.
La evaluación funcional motora se realizó mediante escalas específicas según el tipo de AME: CHOP INTEND y HINE-2 para AME tipo 1; HFSME y RULM para AME tipo 2. Las (Figuras 1 y 2) ilustran la evolución de los pacientes AME tipo 1 en CHOP INTEND y HINE-2, respectivamente, a lo largo del seguimiento. La (Figura 3) compara los valores observados de CHOP INTEND con la evolución esperada en la historia natural de la enfermedad (modelo de Finkel y colaboradores, 2014)7. En la (Tabla 3) se detallan las diferencias individuales respecto a esa trayectoria. En los pacientes AME tipo 2, las (Figuras 4 y 5) muestran la evolución según HFSME y RULM. Las (Figuras 6 y 7) comparan las trayectorias observadas versus las estimadas para la evolución natural de la enfermedad, ajustadas al tiempo de evolución individual16. Las (Tablas 4 y 5) resumen las medidas de tendencia central y dispersión para estas escalas.

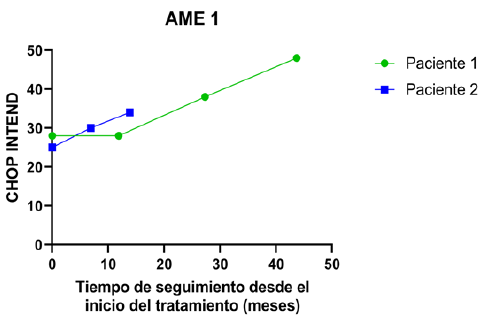
Figura 1 CHOP INTEND en relación con el tiempo de tratamiento. Se presentan los valores de la escala CHOP INTEND para los pacientes AME 1.
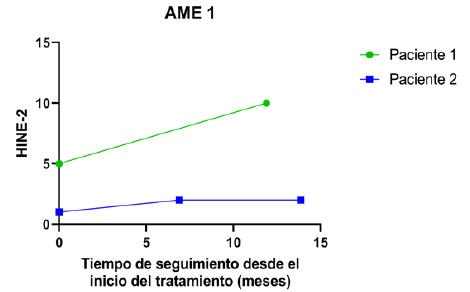
Figura 2 HINE-2 en relación con el tiempo de tratamiento. Se presentan los valores de la escala HINE-2 para los pacientes AME 1.
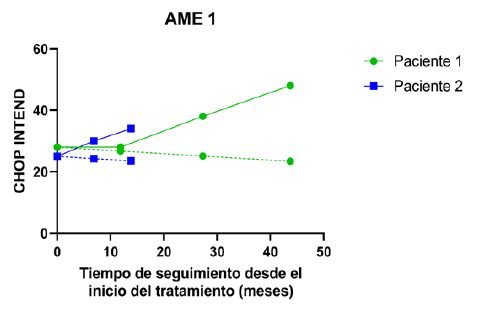
Figura 3 Trayectorias observadas y esperadas del CHOP INTEND en relación con el tiempo de tratamiento. Se presentan los valores observados de la escala CHOP INTEND (línea continua) para los pacientes AME 1 y sus correspondientes valores esperados (línea discontinua) de acuerdo a lo que sería la progresión natural de la enfermedad.
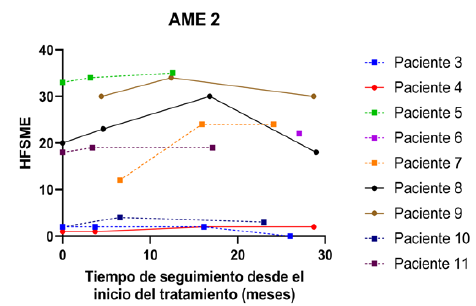
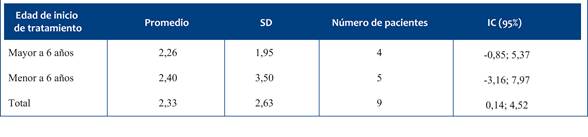
Figura 4 HFSME en relación con el tiempo de trata miento. Se presentan los valores observados de la escala HFS ME para los pacientes AME 2. Se diferencia a los pa cientes menores de 6 años al momento del inicio del tratamiento (línea discontinua y símbolo cuadrado) de los pacientes mayores de 6 años (línea continua y símbolo circular).
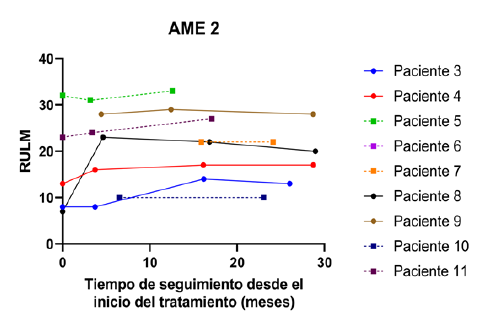
Figura 5 RULM en relación con el tiempo de tratamiento. Se presentan los valores observados de la escala RULM para los pacientes AME 2. Se diferencian a los pacientes menores de 6 años al momento del inicio del tratamiento (línea discontinua y símbolo cuadrado) de los pacientes mayores de 6 años (línea continua y símbolo circular).
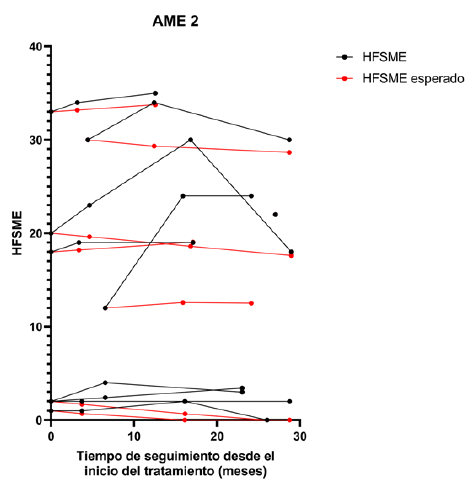
Figura 6 Trayectorias observadas y esperadas del HFSME en relación con el tiempo de tratamiento. Se presentan los valores observados de la escala HFSME (negro) para los pacientes AME 2 y sus correspondientes valores esperados (rojo) de acuerdo a lo que sería la progresión natural de la enfermedad.
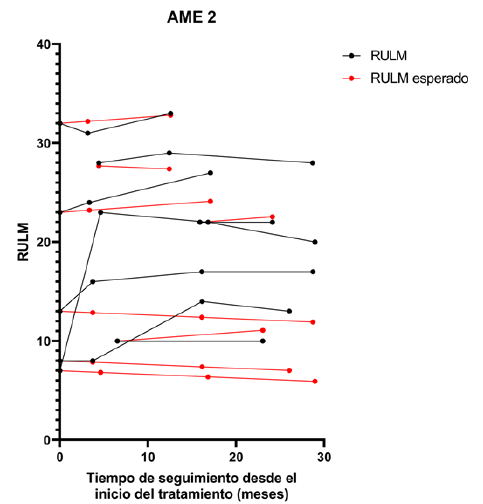
Figura 7 Trayectorias observadas y esperadas del RULM en relación con el tiempo de tratamiento. Se presentan los valores observados de la escala RULM (negro) para los pacientes AME 2 y sus corres pondientes valores esperados (rojo) de acuerdo a lo que sería la progresión natural de la enfermedad.
Discusión
El presente estudio tiene como objetivo analizar la respuesta terapéutica de un grupo de pacientes con AME que iniciaron el tratamiento con nusinersen en Uruguay. Al ser el primer trabajo de este tipo realizado en el país, no contamos con datos comparativos previos para contextualizar los resultados obtenidos. Este estudio presenta algunas limitaciones metodológicas derivadas de su diseño. En primer lugar, la muestra es no probabilística y se obtuvo por conveniencia. Además, no se profundizó en las razones detrás del retraso diagnóstico ni en los factores que incidieron en el cumplimiento del tratamiento. Por otro lado, al tratarse de una enfermedad rara, el tamaño de la muestra fue pequeño, lo que limita la posibilidad de extrapolar los resultados a poblaciones más amplias.
En cuanto a las características de la población de estudio, esta muestra es homogénea en términos de sexo, con 5 niños y 6 niñas. Aunque se trata de una muestra pequeña, con solo 11 pacientes (2 diagnosticados con AME tipo 1, 18,2%, y 9 con AME tipo 2, 81,8%), los datos obtenidos muestran que la edad de presentación de los síntomas en los pacientes con AME tipo 1 fue antes de los 6 meses de vida, mientras que en los pacientes con AME tipo 2, los síntomas aparecieron antes de los 18 meses de edad. Además, los hitos motores alcanzados fueron el sostén cefálico en los pacientes con AME tipo 1 y la bipedestación en los pacientes con AME tipo 2. Estos resultados son consistentes con lo reportado en la bibliografía existente sobre la enfermedad1,6,8,10,11.
La edad de confirmación genética presentó una notable variabilidad en nuestra muestra, con una media de 27,5 meses y una moda de 11 meses. Llama particularmente la atención que dos pacientes con AME tipo 2 recibieron el diagnóstico de forma significativamente tardía, a los 84 y 72 meses de vida respectivamente. Esta situación pone de relieve la necesidad de fortalecer la sospecha clínica frente a signos neuromusculares sutiles, como la hipotonía persistente, la debilidad muscular axial o proximal de inicio temprano y la dificultad para alcanzar hitos motores esperados para la edad, como el sostén cefálico, la sedestación o la marcha1. Resulta fundamental promover el acceso oportuno a estudios genéticos confirmatorios desde el primer nivel de atención, así como fomentar el conocimiento de estas patologías entre pediatras generales y médicos de familia, con el objetivo de reducir los tiempos diagnósticos.
En cuanto a la técnica utilizada, la confirmación genética de la enfermedad se realizó exclusivamente mediante MLPA. El hallazgo más frecuente fue la deleción en homocigosis del exón 7 del gen SMN1 (90,9%). Solo 1 paciente (9,1%) presentó una alteración diferente, consistente en una deleción en homocigosis del exón 5 del gen NAIP y una deleción en heterocigosis de los exones 1, 4, 6 y 8 del gen SMN1. Estos hallazgos son compatibles con los informes previos en la literatura4-6. En cuanto al número de copias del gen SMN2, un paciente con AME tipo 1 presenta dos copias, mientras que el otro tiene tres. Todos los pacientes con AME tipo 2 cuentan con tres copias del gen SMN2, lo que es coherente con la relación genotipo-fenotipo descrita en trabajos anteriores4,8.
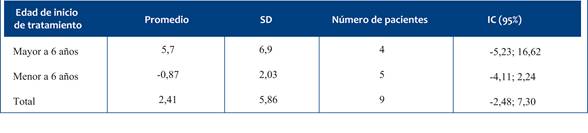
En relación con las escalas de valoración motora, dividiremos a los pacientes según su diagnóstico de AME tipo 1 y AME tipo 2, tal como se presenta en los resultados. En nuestra muestra, los niños con AME tipo 1 que recibieron nusinersen muestran una mejoría en las evaluaciones motoras individuales y en comparación con las trayectorias esperadas para la evolución natural de la enfermedad7, a pesar de continuar requiriendo asistencia ventilatoria. Sin embargo, es importante señalar que estos resultados no tienen suficiente poder estadístico para detectar diferencias significativas que puedan replicarse en otras poblaciones debido al tamaño reducido de nuestra muestra (n=2). Estos hallazgos reflejan una respuesta clínica heterogénea al tratamiento, fenómeno ampliamente documentado en estudios internacionales. La variabilidad puede estar influida por factores como la edad de inicio del tratamiento, el número de copias del gen SMN2, el nivel de compromiso basal, así como por el acceso y adherencia a terapias de rehabilitación y apoyo psicosocial complementario16-19.
En los niños con AME tipo 2 se observó, en general, una evolución funcional estable o con discreta mejoría tras el tratamiento con nusinersen. Esta tendencia resulta especialmente relevante si se tiene en cuenta que la evolución natural de la enfermedad es a la pérdida de habilidades motoras. Al analizar las trayectorias de forma segmentada según la edad de inicio del tratamiento, no se identificaron diferencias estadísticamente significativas en la escala HFSME ni en la RULM. Sin embargo, al considerar el comportamiento global de la cohorte, los resultados sugieren un efecto clínicamente favorable, con pacientes que mantuvieron o incluso superaron levemente lo esperado en función de su estado inicial.
Este hallazgo cobra valor desde la práctica clínica, aun en ausencia de significación estadística, ya que la estabilidad funcional en una patología de curso progresivo, como la AME tipo 2, puede ser interpretada como una respuesta terapéutica beneficiosa. Es probable que el tamaño muestral reducido haya limitado la potencia del análisis para detectar diferencias formales entre subgrupos, pero los datos disponibles permiten plantear una interpretación positiva en términos de impacto funcional global.
Cabe destacar el caso del paciente 9, quien presentó una disminución abrupta en su puntuación motora. A diferencia del resto de la cohorte, su evolución parece haber estado condicionada por factores extraneurológicos, como obesidad y síntomas depresivos, que interfirieron con la adherencia al tratamiento y al abordaje rehabilitador. Este caso subraya la importancia de considerar el contexto psicosocial y los factores de comorbilidad al interpretar los resultados funcionales individuales.
A partir de estos resultados, surge también la necesidad de reflexionar cuidadosamente sobre la indicación de nusinersen en pacientes con estadios avanzados, en particular aquellos que inician el tratamiento de forma tardía, como ocurre con la mayoría de los niños con AME tipo 2 incluidos en esta cohorte. Diversos estudios han demostrado que la eficacia del tratamiento es significativamente mayor cuando se administra en fases presintomáticas o en etapas muy tempranas de la enfermedad, lo que resalta la importancia del tiempo como factor determinante en la respuesta terapéutica. En este sentido, aunque se observen beneficios funcionales globales, es fundamental avanzar hacia estrategias que permitan identificar y tratar a los pacientes en forma más precoz20. Desde una perspectiva bioética, esta situación plantea interrogantes sobre la proporcionalidad del tratamiento y la justicia distributiva, especialmente en contextos donde los recursos son limitados y el acceso a terapias de alto costo puede estar condicionado. Es fundamental considerar no solo la potencial eficacia individual, sino también los principios de equidad y uso racional de recursos sanitarios en la toma de decisiones clínicas y políticas21.
En la actualidad, además del nusinersen, Uruguay cuenta con acceso a otras estrategias terapéuticas, como risdiplam, un modulador oral del splicing del gen SMN2, aprobado para su uso en diversos tipos de AME. También se encuentra disponible la terapia génica con onasemnogene abeparvovec en algunos países. En paralelo, se han iniciado estudios que exploran la posibilidad de ajustar la posología de nusinersen en función de variables como la edad del paciente, la evolución funcional o ciertos marcadores genéticos, con el objetivo de optimizar la eficacia del tratamiento en subgrupos específicos22.
Conclusiones
La AME cromosoma 5q es una enfermedad genética rara, con una incidencia estimada de 1 en 10.000 nacidos vivos y una prevalencia de 1 a 2 por cada 10.000 personas. Es la causa monogénica más frecuente de mortalidad infantil22 y se caracteriza por una notable heterogeneidad tanto en su presentación clínica como en la respuesta al tratamiento.
Los tratamientos modificadores de la enfermedad, como el nusinersen -fármaco objeto de este estudio- han demostrado resultados alentadores en múltiples cohortes clínicas, lo que subraya la importancia de un diagnóstico oportuno y un inicio precoz de la terapia para maximizar su efectividad.
En nuestra serie, se observa una variabilidad en la respuesta terapéutica, con mejorías en las escalas de evaluación motora, aunque sin alcanzar significancia estadística, probablemente debido al tamaño reducido de la muestra. Esta respuesta no es uniforme ni lineal: se evidencian fluctuaciones y una distribución heterogénea de los efectos según los grupos musculares. Si bien se documentan avances en la función motora, la persistencia de requerimientos ventilatorios sugiere que los músculos bulbares responden de manera diferente al tratamiento en comparación con los músculos axiales o de las extremidades.
Por otro lado, la mayoría de nuestros pacientes con AME tipo 2 iniciaron tratamiento en forma tardía, en el contexto de cohortes “históricas”. Esta situación refuerza la premisa de que los fármacos modificadores de la enfermedad detienen la pérdida de motoneuronas, pero no promueven su regeneración. En consecuencia, cuanto más temprano se inicia el tratamiento, mayor es el potencial de beneficio clínico. Además, el aumento en la expectativa de vida asociado al tratamiento ha dado lugar a un nuevo perfil fenotípico, con desafíos terapéuticos y sociales emergentes que merecen seguimiento continuo.
En este contexto, la inclusión de la AME en los programas de pesquisa neonatal adquiere un rol fundamental. La evidencia ha demostrado que los mejores resultados clínicos se alcanzan cuando el tratamiento se inicia en pacientes presintomáticos23. Estados Unidos, Alemania y Australia ya han incorporado la pesquisa neonatal para AME dentro de sus paneles nacionales, basándose en criterios de costo-efectividad, carga de enfermedad y disponibilidad terapéutica24,25. Uruguay cuenta actualmente con cobertura del tratamiento a través del FNR, lo que facilita el acceso y permitiría una implementación efectiva de un programa de pesquisa ampliada. Su incorporación también permitiría detectar casos antes del inicio de los síntomas y maximizar los beneficios del tratamiento.
Finalmente, se considera necesario ampliar este estudio a fin de evaluar en profundidad las complicaciones asociadas con la AME y su impacto en la calidad de vida, tanto de los niños como de sus familias, mediante el uso de herramientas validadas para este fin.

