











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Introducción
Las patologías desmielinizantes son un espectro de enfermedades inmunomediadas que afectan al sistema nervioso central dentro de las que se destacan: esclerosis múltiple, encefalomielitis diseminada aguda (ADEM), neuritis óptica (ON), mielitis transversa longitudinalmente extensa (MTLE) y espectro de neuromielitis óptica (NMOSD). Estas enfermedades son un desafío diagnóstico, ya que pueden evolucionar de un fenotipo a otro, pudiendo presentar diversidad de manifestaciones clínicas según el área afectada. Actualmente existen anticuerpos específicos que funcionan como biomarcadores útiles para identificar la etiología en cada caso1-4.
Los anticuerpos contra la glucoproteína de la mielina de los oligodendrocitos (Ac-antiMOG) se dirigen a MOG, el cual representa solo el 0,5% de la mielina y se expresa en la membrana plasmática de los oligodendrocitos, siendo muy específica e inmunogénica5-7.
Los Ac-antiMOG generan un espectro de síndromes desmielinizantes muy frecuentes en la edad pediátrica, configurando en sí mismos una entidad propia e independiente nombrada enfermedad asociada a anticuerpos MOG (MOGAD).
El tratamiento se basa en inmunoterapia, en los eventos agudos se indica escalonadamente el uso de corticoides endovenosos, plasmaféresis e inmunoglobulina intravenosa (IGIV), el tratamiento de mantenimiento crónico debe ser individualizado en cada caso, pudiendo usarse glucocorticoides orales (GCO), azatioprina (AZA), micofenolato de mofetilo (MMF), inmunoglobulinas (IGIV) y rituximab (RTX)1.
El pronóstico generalmente es favorable, presentando una recuperación completa en la mayoría de los casos. Sin embargo, esta evolución depende de la severidad de la presentación inicial y las recaídas8,9.
La justificación teórica en la que se engloba este trabajo se basa en la ausencia de estudios en el ámbito nacional que describan esta patología. El objetivo es señalar las características clínicas, los tratamientos realizados y la evolución de un grupo de niños con diagnósticos de MOGAD atendidos en el Servicio de Neuropediatría del Centro Hospitalario Pereira Rossell (CHPR), entre enero de 2022 y junio de 2024.
Metodología
Se trata de un estudio observacional, descriptivo, retrospectivo, tipo reporte de casos, basado en la revisión de las historias clínicas de todos los pacientes con criterios diagnósticos de MOGAD atendidos en el Servicio de Neuropediatría del CHPR, entre enero de 2022 y junio de 2024. Se analizaron las manifestaciones clínicas, resultados de estudios paraclínicos y la neuroimagen.
Resultados
Se incluyeron 14 pacientes diagnosticados con MOGAD con clínica y resonancia magnética (RM) (Figura 1) compatible con un síndrome desmielinizante agudo y Ac-antiMOG positivos. Para la búsqueda de anticuerpos se utilizaron ensayos basados en células (CBA) prefijados en suero. En todos los casos el estudio de neuroimagen utilizado fue la RM de cráneo con enfoque de órbita y médula total con contraste. Además, se descartaron otras causas infecciosas, así como otras etiologías desmielinizantes.
Los fenotipos clínicos de presentación fueron MTLE, espectro NMOSD, encefalitis, ADEM y síndrome neurológico monofocal. Un niño presentaba obesidad como antecedente personal y otra niña hipoacusia y cataratas congénitas (caso 2 y caso 14, respectivamente). Cuatro niños cursaron infecciones banales semanas previas al debut, uno recibió inmunización para HPV 20 días previo al debut. Todos los pacientes que recibieron tratamiento utilizaron de primera línea metilprednisolona intravenosa (MTIV), cuatro pacientes requirieron tratamientos de segunda línea dada la poca respuesta al tratamiento inicial, todos los niños continuaron con GCO con descenso paulatino. Uno de los niños presentó un brote, reinstalando los síntomas en contexto de descenso de GCO, cuatro pacientes presentaron recaída, en estos casos se realizó nuevo ciclo de MTIV con buena respuesta, continuando luego con GCO. Los pacientes con enfermedad recurrente se mantuvieron con AZA o rituximab. En un paciente con clínica severa al debut también se inició rituximab de mantenimiento, sin recaídas a la fecha. Trece pacientes presentaron una recuperación completa luego del primer evento, un paciente presentó una lenta recuperación (fenotipo MLTE) persistiendo a los seis meses con una paresia leve distal simétrica. En las (Tablas 1,2,3,4) se realiza la descripción sintetizada de cada caso, incluyendo información complementaria y comparativa.
Tabla 1 Resumen fenotipo clínico, exámenes complementarios, tratamiento y evolución de niños con fenotipo MTLE. MTLE: mielitis transversa longitudinalmente extensa; PEV: potenciales evocados visuales; MTIV: metilpred-nisolona intravenosa; IGIV: inmunoglobulina intravenosa.
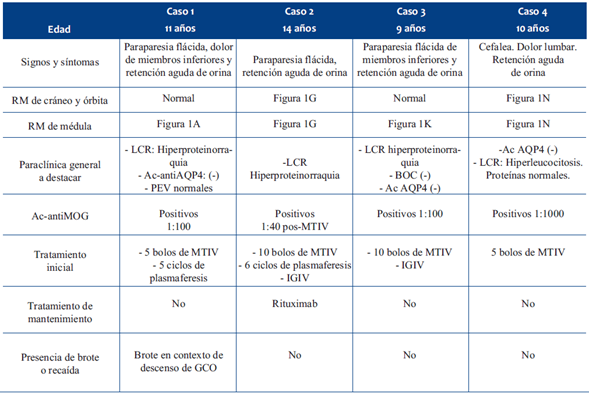
Tabla 2 Resumen fenotipo clínico, exámenes complementarios, tratamiento y evolución de niños con fenotipo NMOSD. NMOSD: espectro de neuromielitis óptica; PEV: potenciales evocados visuales; MTIV: metilprednisolona intravenosa; IGIV: inmunoglobulina intravenosa.
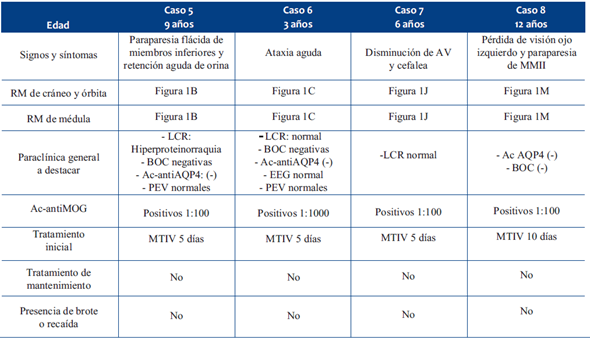
Tabla 3 Resumen fenotipo clínico, exámenes complementarios, tratamiento y evolución de niños con fenotipo encefalitis. ADEM: encefalomielitis diseminada aguda; PEV: potenciales evocados visuales; MTIV: metilprednisolona intravenosa; IGIV: inmunoglobulina intravenosa.
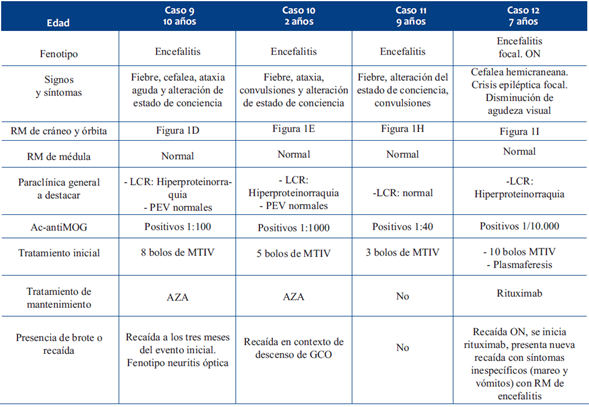
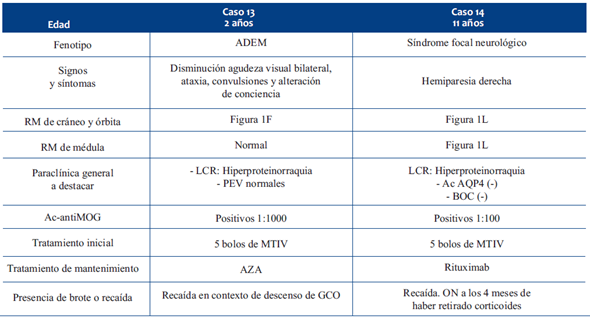
Discusión
Recientemente ha surgido gran interés en el papel de los Ac-antiMOG como marcador de enfermedades desmielinizantes del sistema nervioso central (SNC) dada su alta especificidad (hasta 98% - 100%) e inmunogenicidad5,10. Esta entidad presenta una mayor incidencia en pacientes pediátricos, ocupando el 40% de los casos de síndromes desmielinizantes en este grupo etario versus 20% en población adulta11.
Clásicamente se describen fenotipos clínicos MOGAD tales como: ADEM en 45%, ON en 30%, MTLE en 11% y fenotipo similar a NMOSD 4,5%. Más recientemente se han descrito otros fenotipos, como la encefalitis, leucodistrofias-like, desmielinización central y periférica combinada y fenotipos no clasificables, estos últimos representan el 9,5% de los casos1. La patología tiene como base un mecanismo inmunomediado dado por la presencia de los anticuerpos mencionados, pero se desconoce el mecanismo gatillador, estimándose cualquier entidad que desencadena una cascada inflamatoria exacerbada, destacándose las infecciones virales.
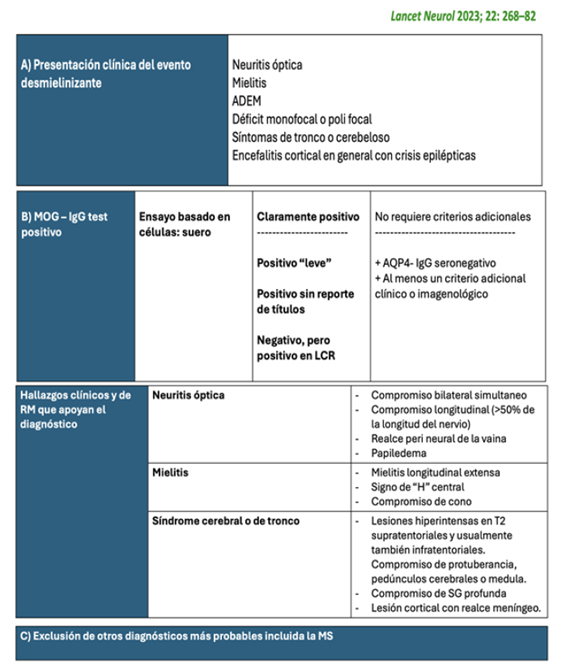
Presentamos 14 pacientes con edades comprendidas entre 2 y 14 años. Como posible mecanismo gatillador, en 6/14 pacientes existió un proceso infeccioso en al menos dos semanas previas al inicio del MOGAD, y en uno actualización del esquema de vacunación, situación similar a la reportada en otras publicaciones, donde las casuísticas identifican un pródromo infeccioso en un tercio de los casos1,12. En cuanto a las manifestaciones clínicas el fenotipo fue variado, 4 tuvieron encefalitis, 4 NMOSD, 4 MTLE, 1 un fenotipo monofocal y 1 ADEM. Está descrito que el fenotipo predominante en niños en MOGAD es el ADEM en 45,6%, seguido de ON 29,6%, MTLE 11,2%, NMOSD 4,3% y otros fenotipos con 9,5%1. Los criterios diagnósticos de MOGAD se describen en la (Tabla 5).
Ocho casos presentaron hiperproteinorraquia, hallazgo habitual junto con pleocitosis en los síndromes desmielinizantes, el cual puede ser más marcado en MOGAD1,13. En los pacientes con fenotipo MTLE y similar NMOSD se buscó como diagnóstico diferencial los anticuerpos contra acuaporina 4 (Ac-anti AQP4), los cuales fueron negativos. En los pacientes en los que se buscaron BOC, fueron negativas, estando descrita la positividad en MOGAD entre 7%-11%14. En cuanto a las técnicas para la determinación de Ac-antiMOG, éstas han evolucionado en los últimos años. Actualmente se buscan anticuerpos contra epítopos conformacionales mediante CBA en células prefijadas a nivel comercial y vivas en centros de investigación, siendo estas últimas más sensibles y específicas. El suero es la muestra de elección para el estudio de MOG, dado que la producción del anticuerpo es de origen sistémico y no intratecal, por lo que la búsqueda del anticuerpo en LCR, en general, arroja resultados negativos. Se consideran positivos títulos en suero mayores a 1:160 en CBA por inmunofluorescencia (IF)13,14. En nuestros pacientes la determinación de anticuerpos se realizó por citometría de flujo (FACS) en CBA prefijadas. En una reciente publicación se demostró que los valores iguales o mayores a 1:100 obtenidos por FACS CBS prefijadas son comparables a los estudios con IF15; en esta serie la mayoría de los casos cumplieron con este criterio de positividad; en los casos 7 y 8, en donde el resultado fue menor a este valor (positivo débil), se confirmó el diagnóstico por cumplir criterios secundarios establecidos en el consenso 202316-21.
En la neuroimagen se observan imágenes inflamatorias, hiperintensas en T2 y FLAIR, el realce con gadolinio en MOGAD es menos frecuente y, si está presente, a menudo es más difuso que en EM o en enfermedad por AQP4. En ADEM se presenta con lesiones grandes y mal delimitadas de sustancia blanca supra y/o infratentoriales (Figura 1 F). En ON se presenta frecuentemente con afectación bilateral, extensa y a predominio anterior del nervio óptico, a diferencia de AQP4 donde suele haber compromiso del quiasma, y de EM donde la afectación suele ser unilateral y corta. La MTLE se caracteriza por lesiones en tres o más segmentos medulares continuos, afectación del cono medular y compromiso central con signo de la H, tal como lo observamos en tres de nuestros pacientes (Figura 1 A, B, C y G); a diferencia de lo que se observa en la enfermedad por AQP4, donde a nivel encefálico se presentan lesiones en la zona periventricular, sustancia gris periacueductal y zona del tronco adyacente al cuarto ventrículo siguiendo un patrón en las áreas ricas en receptores de AQP41,16. Por su parte, en la EM son más comunes las lesiones múltiples, de segmentos cortos y con compromiso lateral1,16. Últimamente se ha descrito un nuevo fenotipo que se presenta con un cuadro de encefalitis que afecta predominantemente la sustancia gris cortical con o sin afectación gangliobasal y talámica simétrica o asimétrica1,13,16, de esta última presentamos cuatro pacientes (Figura 1 D, E, H, I).
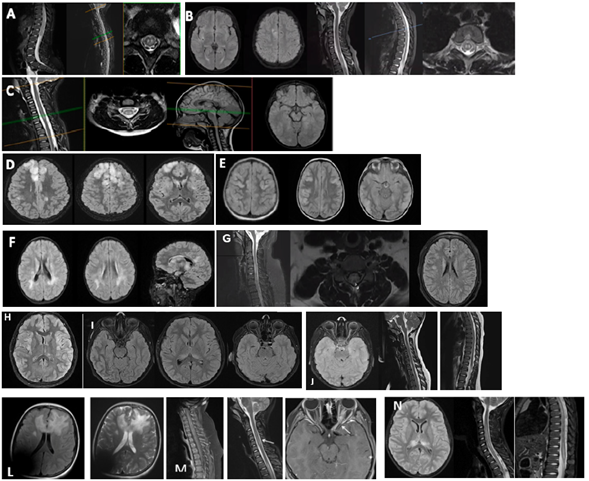
Figura 1 Hallazgos en resonancia magnética: A) RM médula, corte sagital, secuencia T2, hiperintensidad de T3 al cono medular y signo de la H en corte axial. B) RM cráneo, corte axial, secuencia FLAIR, lesiones hiperintensas en sustancia blanca supratentorial. RM medular, corte sagital en T2, hiperintensidad desde t1-t6 y cono medular con signo de la H en corte axial. C) RM médula, corte sagital, secuencia T2, hiperintensidad C5-T1 con signo de la H en corte axial. RM cráneo, corte sagital y axial, secuencia FLAIR, múltiples lesiones hiperintensas córtico-subcorticales, gangliobasales y de tronco encefálico. D) RM cráneo, corte axial, secuencia FLAIR, áreas parcheadas mal definidas hiperintensas a nivel de cortical frontal, núcleo lenticular, caudado e insular bilateral. E) RM cráneo, secuencia FLAIR, corte axial, áreas parcheadas mal definidas hiperintensas a nivel de cortical frontoparietal bilateral. F) RM cráneo, cortes axiales y sagital, secuencia FLAIR, áreas mal definidas hiperintensas en la sustancia blanca supraten torial frontoparietal bilateral y ganglio basal a predominio derecho. G) Lesión a nivel cervical entre los niveles C4 - C5 a C6 - C7, de alta señal en T2 con ensanchamiento medular asociado. RM cráneo secuencia FLAIR múltiples lesiones focales de la sustancia blanca supratentorial, frontoparietales bilaterales. H) RM cráneo en secuencia T2 FLAIR, hiperintensidad bitemporal. I) RM cráneo, secuencia T2 FLAIR, hiperintensidad cortical frontoparietal derecha. Hiperintensidad bitalámica. Hiperinten sidad a nivel de nervio óptico izquierdo. J) Protrusión en ambas pupilas ópticas, aumento de señal en T2 y realce de nervio óptico izquierdo desde el sector retro bulbar hasta su porción intracraneana. Médula: áreas de aumento de señal en T2 que se extienden desde C3 - C4 a C6 - C7. K) Aumento de la señal en T2 a nivel del cordón medular cervical, parcheadas, de longitud intermedia, se extiende desde C3 - C4 a C6 - C7. L) Estudio limitados por artificios. Hiperintensidad T2 y flair bifrontal y sector dorsal de protuberancia, con compromiso de cuerpo calloso. Confluente y mal delimitado. No restringe en difusión. Realce medular C4 - C5 y cola de caballo. M) Secuencia T1 con gadolinio muestra realce del nervio óptico izquierdo. STIR de columna cérvico-dorsal muestra aumen to de señal de varios segmentos vertebrales. N) Hiperintensidad en T2/flair bitalámicas. Lesión medular longitudinalmente extensa con distribución central y compromi so de cono terminal.
En cuanto al tratamiento del MOGAD, en los eventos agudos la inmunoterapia se basa en el uso escalonado de MTIV, plasmaféresis e IGIV, considerando posteriormente el uso de GCO (Figura 2)17. En esta serie, el 100% recibió MTIV, cinco pacientes requirieron tratamiento de segunda línea optando por IGIV y plasmaféresis. La respuesta al tratamiento del evento agudo se analizó en una cohorte retrospectiva, principalmente de pacientes adultos con MOGAD, en donde el 50% presentaron una recuperación completa y 44% recuperación parcial tras el uso de MTIV18, datos similares se observaron en MOGAD pediátrico19.
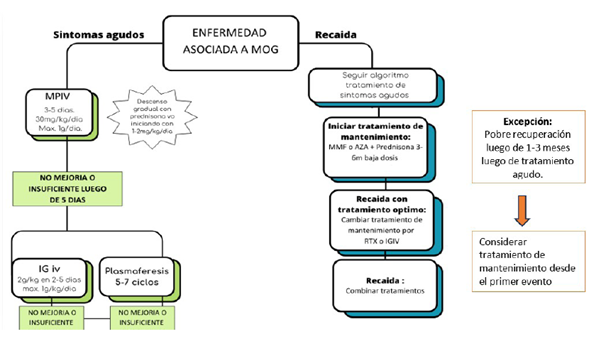
Figura 2 Algoritmo de tratamiento en MOGAD. Adaptación de Bruijstens A, Wendel E, Lechner C, et al. E.U. paedia tric MOG consortium consensus: Part 5 e Treatment of paediatric myelin oligodendrocyte glycoprotein antibody-as sociated disorders, European Journal of Paediatric Neurology.
La presencia de fenotipos recidivantes de MOGAD en pediatría varía de 22% a 60%, siendo el principal factor la persistencia de Ac-antiMOG a largo plazo13. Se define como recaída a un nuevo episodio clínico acompañado de evidencia radiológica según el subtipo de MOGAD, que aparece al menos un mes después del último evento, mientras que brote se definió como la recurrencia de los síntomas dentro del mes del último episodio (hasta tres meses si es ADEM)1. En los casos presentados, un niño tuvo brotes relacionados con el descenso de los GCO, cinco niños tuvieron recaída, con buena evolución posterior.
Las terapias de mantenimiento utilizadas para la prevención de recaídas en MOGAD se detallan en la (Figura 2)17. Estos tratamientos se reservan para casos multifásicos o pacientes con mala respuesta al tratamiento agudo17. En nuestra serie de casos, cuatro pacientes presentaron recaídas, se les indicó tratamiento de mantenimiento con AZA o rituximab, mientras que un paciente recibió rituximab de mantenimiento debido a la presentación clínica severa al debut.
En la bibliografía reportada se destaca que 75% a 96% de los pacientes tienen recuperación completa tras el tratamiento20. En concordancia con esto, en nuestra serie, un único paciente presentó una paresia distal de miembros inferiores como secuela neurológica.
Conclusiones
MOGAD es una enfermedad desmielinizante de mecanismo inmunomediado. Es una entidad emergente, poco prevalente, con un amplio espectro de manifestaciones clínicas que dan origen a diversos fenotipos.
Requiere de un alto índice de sospecha clínica, existiendo un biomarcador específico y sensible: el Ac-antiMOG. Destacamos la importancia de éste, siendo la muestra de elección para su estudio el suero. Actualmente la técnica válida para su búsqueda es mediante CBA con títulos igual o mayores a 1:100 por FACS o < a 1:100, pero asociado a criterios secundarios y negatividad de AQP4.
Su diagnóstico precoz y tratamiento oportuno habitualmente presenta una buena respuesta sin necesidad de terapias de mantenimiento y en la mayoría de los casos sin dejar secuelas neurológicas.

