











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Introducción
Las anomalías cromosómicas son una causa importante de muerte perinatal y discapacidad en la infancia1-3. El síndrome de Down (SD) o trisomía 21 es la alteración cromosómica más frecuente en el ser humano1,2,4, asimismo constituye la principal causa de discapacidad intelectual de origen genético1. Tiene una incidencia general de 1 en 600 a 800 recién nacidos vivos2,5, la cual aumenta en neonatos o fetos de gestantes mayores a 35 años1.
Alrededor del 95% de los pacientes con SD presenta una trisomía libre del cromosoma 211,6-8. El 3%-4% presenta una translocación robertsoniana entre el brazo largo del cromosoma 21 y el brazo largo de uno de los otros cromosomas acrocéntricos, la más frecuente 14;218. En este último caso no existe relación con la edad materna y el riesgo de recurrencia es muy alto, haciendo necesaria la realización de un cariotipo a ambos progenitores1,5. Finalmente, el 1%-2% de los pacientes con SD son mosaicos1,2,8.
Los pacientes con SD presentan un fenotipo característico1. En el recién nacido la hipotonía es el signo cardinal en el diagnóstico, presentándose en el 80% de los pacientes6. Ciertos defectos congénitos, como las cardiopatías (presente en el 50% de los casos8-10), la atresia duodenal, la enfermedad de Hirschsprung y/o la fístula traqueoesofágica aislada, son más frecuentes en estos recién nacidos1,11, con un impacto directo en la morbimortalidad de los pacientes. El defecto más común a nivel cardiovascular es la comunicación interventricular (CIV) perimembranosa, seguida por la comunicación interauricular (CIA) y el canal atrioventricular común (canal AV)8. El 80% de los canales AV corresponden a pacientes con SD, siendo altamente sugestivo del mismo cuando se diagnostica de forma prenatal; no obstante, el 70% de los pacientes con trisomía 21 presenta como cardiopatía más frecuente la CIV8,11. Otras comorbilidades frecuentes que se describen en estos pacientes son las alteraciones endocrinológicas, como el hipotiroidismo1,8, así como alteraciones hematimétricas benignas y malignas. Dentro de las primeras se destacan: neutrofilia (80%), plaquetopenia (66%) y policitemia (64%)12,13.
El SD puede ser diagnosticado prenatalmente3,8, permitiendo planificar el lugar y el momento del nacimiento, preparar a la familia y así mejorar los resultados perinatales1. El estudio citogenético a través de la realización de una biopsia de vellosidades coriónicas, entre las semanas 9 y 12, o por amniocentesis entre las semanas 14 y 208, es un procedimiento invasivo que implica riesgos para el feto. Es por ello que se han desarrollado diferentes pruebas de screening, no invasivas, que estiman riesgo pero no diagnostican que el feto tenga SD. Las pruebas de cribado son sencillas, inocuas, ofrecen datos inmediatos y permiten reservar la realización de las pruebas invasivas solo para los casos con alto riesgo1,3. El tamizaje combinado bioquímico-ecográfico del primer trimestre, que incluye la cuantificación de la proteína A del plasma sanguíneo asociada al embarazo (PAPP-A), la subunidad beta libre de la gonadotropina coriónica humana (fβHCG) y medición de la translucencia nucal por ecografía alcanzan una sensibilidad de detección de 90%-95%3. En el segundo trimestre se puede realizar el análisis bioquímico de alfa-fetoproteína (AFP), la gonadotropina coriónica humana total (HCGt), el estriol no conjugado (uE3), conocido como “prueba triple” entre las semanas 15 y 18, o “prueba cuádruple” si se adiciona inhibina A, con un porcentaje de detección de 85% y 90%, respectivamente. Un resultado normal reduce la probabilidad SD, pero no lo excluye. Existen otros hallazgos ecográficos asociados al SD, como la ausencia del hueso nasal, higroma quístico, la presencia del ductus venoso y la presencia de regurgitación en la válvula tricúspide del corazón, entre los más relevantes. Otra técnica alternativa es el test prenatal no invasivo, conocido como ADN fetal de células libres en sangre materna (cfADN: cell-free ADN), con una sensibilidad de 98%-99% para detección de SD3. Puede realizarse desde las nueve semanas de gestación.
El diagnóstico posnatal es de sospecha clínica1, basado en las características fenotípicas8. El estudio de cariotipo es necesario para confirmar el diagnóstico y realizar un adecuado asesoramiento genético1,2. Al momento de asesorar a una familia es importante conocer que la incidencia del SD en recién nacidos vivos es de 1/600 a 1/800, lo que refleja la distribución de la edad materna en la población general1, y que el riesgo comienza a aumentar a partir de los 35 años para la trisomía libre. El riesgo de SD por translocación o mosaicismo no está relacionado con la edad materna. El riesgo de recurrencia de trisomía 21 tras el nacimiento de un niño con SD es de aproximadamente 1%1. Los antecedentes de SD en la familia, que no sea un hijo previo, no aumentan el riesgo. Finalmente, el riesgo de recurrencia de SD por translocación es muy alto.
Existen pocos estudios que describan neonatos uruguayos con SD, por lo que resulta de gran importancia caracterizarlos y conocer sus comorbilidades con el objetivo de poder elaborar herramientas de trabajo y políticas de salud que nos permitan mejorar la calidad de atención a estos recién nacidos, haciéndola más adecuada y oportuna. El objetivo de este estudio fue describir las características fenotípicas, genéticas, defectos congénitos y morbilidades asociados en los pacientes con SD nacidos en el Servicio de Neonatología del Centro Hospitalario Pereira Rossell en el período comprendido entre el 1 de julio de 2017 y el 1 de julio de 2021.
Metodología
Se realizó un estudio descriptivo en donde fueron incluidos todos los recién nacidos con diagnóstico de SD, nacidos y/o hospitalizados en el Servicio de Neonatología del Centro Hospitalario Pereira Rossell en el período comprendido entre el 1 de julio de 2017 y el 1 de julio de 2021.
Como variables se analizaron las características del nacimiento, clasificación de los recién nacidos (sexo, edad gestacional, peso al nacer, peso para la edad gestacional), características fenotípicas y asociadas a morbilidad, en donde se incluyeron anomalías congénitas a nivel cardiovascular, digestivo y del sistema nervioso central, alteraciones hematológicas benignas e hipotiroidismo. En lo cardiovascular fueron tomados en cuenta todos los hallazgos ecocardiográficos que incluían cardiopatías estructurales. No se incluyó el ductus arterioso permeable en pretérminos, ya que se valoró como asociados a la prematurez. Otras variables medidas fueron estancia hospitalaria, mortalidad y resultado de cariotipo.
La recolección de datos se hizo utilizando el sistema informático perinatal (SIP) y la historia clínica electrónica a través de la plataforma GeoSalud. Se recogieron los datos de los pacientes en una tabla estandarizada, diseñada por los autores y anonimizada. Este estudio fue aprobado por el Comité de Ética del Centro Hospitalario Pereira Rossell - Hospital de la Mujer.
Resultados
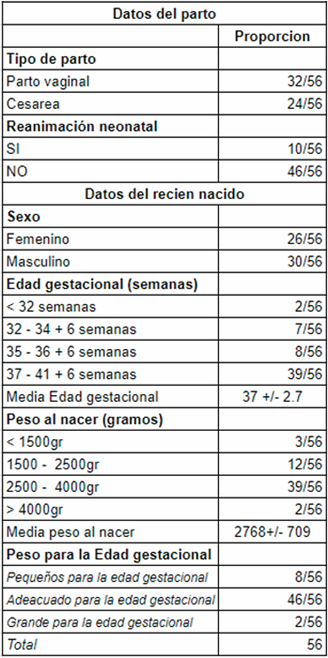
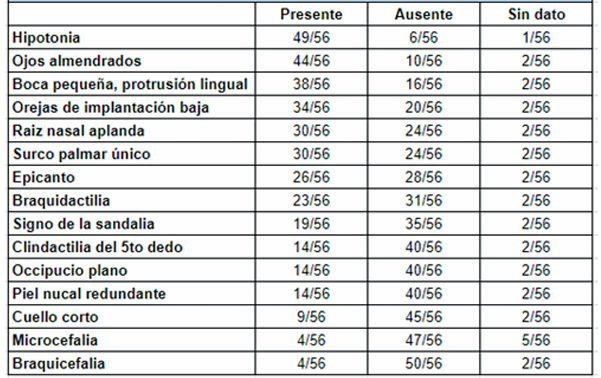
Se incluyeron en el estudio un total de 56 pacientes con diagnóstico de SD, sobre un total de 24.102 recién nacidos vivos en el período analizado. De los pacientes estudiados, 4/56 tenían diagnóstico prenatal de SD por cariotipo. En la (Tabla 1) se encuentra la descripción general de la población de estudio. Treinta fueron de sexo masculino. La media de edad gestacional fue de 37 semanas +/- 2,7. Un total de 17 pacientes fue prematuro. Treinta y nueve pacientes fueron normopeso, con una media de 2.768 g al nacer +/- 709 g. Del total, 46 tuvieron un peso adecuado para la edad gestacional y 8 fueron pequeños para la edad gestacional. En cuanto a la caracterización genotípica, 45 pacientes presentaron trisomía libre, 10 mosaicismo en diferentes porcentajes y un paciente translocación robertsoniana. En la (Tabla 2) se describen las características fenotípicas. La hipotonía fue el signo más frecuentemente encontrado en el examen clínico, descrito en 49 neonatos. Le siguen en frecuencia algunas dismorfias faciales, tales como ojos almendrados 44/56, boca pequeña con protrusión lingual 38/56, orejas de implantación baja 34/56 y raíz nasal aplanada 30/56. Con menor frecuencia se describen otras dismorfias faciales y de manos y pies. En la (Tabla 3) se describen los defectos congénitos y las comorbilidades por aparatos y sistemas. La cardiopatía congénita fue el defecto congénito más frecuente, encontrándose en 34 de 56 pacientes. La más frecuente fue la CIA en 20 pacientes, la sigue la CIV y el canal AV. Solo cuatro de los 34 pacientes a los que se les diagnosticó cardiopatía congénita al nacimiento, tenían diagnóstico prenatal. Por otra parte, siete pacientes presentaron defectos congénitos gastrointestinales y tres del sistema nervioso central.
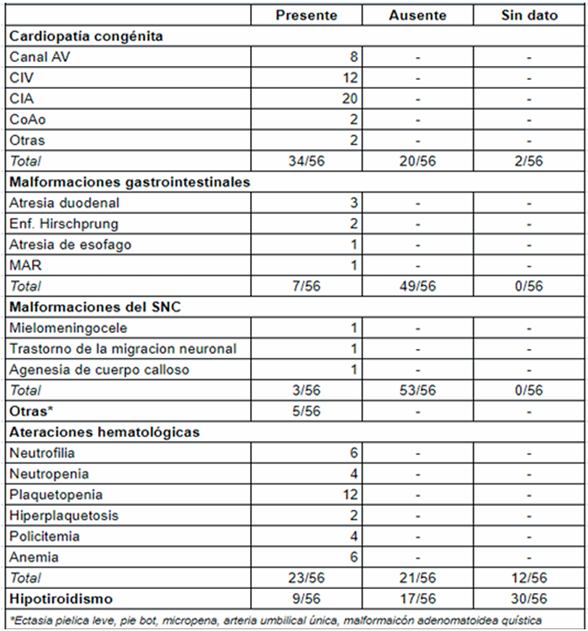
Se encontraron 23 pacientes con alteraciones en el hemograma del nacimiento y se realizó diagnóstico de hipotiroidismo durante la internación a nueve pacientes, de estos solo tres mostraron un valor de TSH en sangre de cordón alterada. Por último, la media en días de estadía hospitalaria fue de 17, con un rango entre 2 y 66 días. Treinta y un pacientes requirieron ingreso a áreas de internación, incluyendo cuidados moderados, intermedios y críticos. Solo un paciente falleció previamente al alta, en el cual se destaca la prematurez extrema como causa.
Discusión
Respecto a las características generales, en la población de estudio se observó que el sexo masculino fue más frecuente, la misma tendencia se reportó en un estudio realizado en nuestro país por Larrandaburu M14, en la que se usaron los datos del Registro Nacional de Defectos Congénitos y Enfermedades Raras (RNDCER), entre 2011 y 2014, el cual mostró que el sexo masculino fue más frecuente, 57% de los casos, coincidiendo también con lo reportado por Moraes M15 de 53,3% en otra cohorte uruguaya de 45 niños con SD derivados a la Policlínica interdisciplinaria de atención al niño con síndrome de Down (PIANSD). La media de edad gestacional fue de 37 semanas. Un estudio realizado en Holanda por Michel E. Weijerman16, donde se estudiaron 182 niños con SD, mostró una edad gestacional de 38 semanas. Por otro lado, se observó que 17 de 56 pacientes (30%) fue prematuro; Nakousi N17 estudió 140 neonatos chilenos con trisomía 21 y observó un porcentaje similar de prematurez (24,7%). La causa del nacimiento prematuro en estos neonatos podría estar en relación con la presencia de defectos congénitos17. En el presente estudio, a 12 de 17 (70%) pretérminos se les detectó un defecto congénito, similar a Nakousi N17, aunque no se encontró diferencia significativa respecto a los recién nacidos a término. El SD se ha asociado con menor peso al nacer17,18. En este estudio 39 (69%) pacientes fueron normopeso, con una media de 2.768 g al nacer, menor que E. Weijerman16, que reportó 3.119 g en su población de estudio. Del total, 46 (82%) tuvieron un peso adecuado para la edad gestacional y 8 (14%) fueron pequeños para la edad gestacional. Dentro de estos últimos, 6 de 8 pacientes tenían diagnóstico prenatal de restricción del crecimiento intrauterino (RCIU), en cinco de ellos se identificaron patologías maternas de base y propias del embarazo, como el tabaquismo, consumo de sustancias psicoactivas y estado hipertensivo del embarazo en sus diferentes clasificaciones. En un caso no se encontró causa materna de RCIU. En las dos pacientes sin diagnóstico prenatal de RCIU, no se identificaron causas maternas objetivables de RCIU. Solo cuatro pacientes (7%) cumplieron con ambas condiciones, prematurez y ser pequeños para la edad gestacional. Estos datos enfatizan que el bajo peso al nacer no solo está determinado por la propia constitución genética del paciente, sino que también tienen un rol preponderante los factores placentarios y uterinos17.
En cuanto a la caracterización genotípica, encontramos mayor cantidad de recién nacidos mosaicos y menor de trisomía libre en comparación a la bibliografía clásica, que estima un 95%1,6-8,19 de trisomía libre, 3%-4% de translocación robertsoniana1,8,19 y 1%-2% de mosaicismo1,2,8,19. Este último hallazgo podría estar explicado por el escaso número de pacientes que conforman la cohorte. Sin embargo, la edad materna podría estar vinculada, ya que en nuestro estudio 46,5% de las madres tenían menos de 35 años.
Respecto a la caracterización fenotípica los resultados obtenidos concuerdan ampliamente con la bibliografía internacional. La hipotonía fue el signo más frecuentemente encontrado en el examen clínico, junto a las dismorfias faciales características. En una serie de 48 recién nacidos con trisomía 21, todos tenían cuatro o más características, y el 89% tenía seis o más de las siguientes: perfil facial plano, fisuras palpebrales inclinadas, oídos anómalos, hipotonía, pobre reflejo de Moro, displasia de la mitad de la falange del quinto dedo, pliegue transverso palmar, piel excesiva en la nuca del cuello, hiperflexibilidad de las articulaciones, displasia de la pelvis6,20.
Se ha asociado el SD con la presencia de anomalías congénitas, siendo las más frecuentes las cardiopatías18. En este estudio, a 37 de 56 (66%) neonatos incluidos con SD se les detectó algún defecto congénito, menor a lo reportado por Moraes M15 de 80% y similar a Nakoski N17, que describe 70,7%. Otro estudio, realizado por Heinke D21 en Estados Unidos, con datos provenientes de 13.376 niños con SD, entre 2013 y 2017, mostró que 4.662 (75,1%) tenían al menos un defecto congénito. De acuerdo a la bibliografía internacional, aproximadamente la mitad de las personas con SD tienen cardiopatía congénita2,6,22,23. En la población estudiada, 34 pacientes (61%) presentaron cardiopatía congénita, siendo este el defecto congénito asociado más frecuente, similar a Nakoski N17 (64,3%) y algo menor a Moraes M15 (71%). Heinke D21, en su serie, también reportó los defectos cardiovasculares como los más frecuentes: 65,6% de todos los casos. Debemos destacar que dentro de las cardiopatías congénitas la más frecuente fue la CIA, seguida por la CIV en segundo lugar y canal AV en tercer lugar, coincidiendo con lo reportado por Heinke D21 de 32,5% de defectos septales a nivel auricular, 20,6% de defectos septales a nivel ventricular y 17,4% de defectos septales atrioventriculares.
Por otra parte, las alteraciones hematológicas son frecuentes en el SD. Respecto a la plaquetopenia, que fue el hallazgo más común (12 pacientes), en siete de ellos no se encontró causa materna u otra patología perinatal distinta del síndrome genético como causa posible. Los cinco neonatos restantes tenían como antecedente, además del SD, madres con estados hipertensivos del embarazo (EHE) y diagnóstico prenatal de RCIU. Por otro lado, la policitemia se encontró en cuatro recién nacidos, 7% de los casos, bastante menor de lo que reporta la literatura, que estima aproximadamente el 65% de los recién nacidos con trisomía 21, pudiéndose explicar por la hipoxemia fetal crónica24,25. Ninguno de los pacientes presentó reacción leucemoide. Los trastornos tiroideos también son comunes en el SD. La prevalencia varía dependiendo en parte de la población estudiada y la edad de la prueba26. En este estudio se reportaron nueve neonatos (16%) con trisomía 21 e hipotiroidismo. Moraes M15 reportó que la incidencia de hipotiroidismo aumenta desde 2,2% al nacimiento hasta 18,2% en los niños que cumplen un año de seguimiento, por lo que cobra real importancia la realización seriada de estudios de hormona tiroidea, debido a que el resultado normal al nacimiento puede tornarse alterado durante el primer año de vida15.

