










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkArchivos de Pediatría del Uruguay
versión On-line ISSN 1688-1249
Arch. Pediatr. Urug. vol.76 no.3 Montevideo 2005
CASO CLÍNICO
Arch Pediatr Urug 2005; 76(3)
Deficiencia de ornitina transcarbamilasa: presentación de un caso clínico
DRES. ANDREA INCORONATO 1, AíDA LEMES 2, ALFREDO CERISOLA 3,4, FERNANDA PéREZ 3, MARíA ELENA TEJERO 1, ALICIA MONTANO 5
1. Médico Pediatra. Ex-Residente de Clínica Pediátrica “B”.2. Médico. Instituto de Genética Médica, Hospital Italiano, Montevideo.
3. Médico Pediatra. Ex-Asistente de Clínica Pediátrica “B”.
4. Asistente de la Cátedra de Neuropediatría
5. Profesora de Clínica Pediátrica “B”
Instituciones responsables: Clínica Pediátrica “B” Prof. Dra. Alicia Montano. Facultad de Medicina, Universidad de la República, Montevideo, Uruguay. Instituto de Genética Médica. Hospital Italiano. Montevideo. Uruguay
Fecha recibido: 24 de agosto de 2005.
Fecha aprobado: 17 de octubre de 2005.
Resumen
Se presenta el caso clínico de una preescolar de dos años y tres meses con antecedentes de sintomatología neurológica y digestiva recurrentes en la cual el perfil bioquímico es consistente con hiperamoniemia primaria por deficiencia de ornitina transcarbamilasa. La evolución clínica y bioquímica fue satisfactoria al iniciarse el tratamiento. Si bien los síntomas aislados son totalmente inespecíficos, es nuestro interés subrayar la importancia que el pediatra sospeche la posibilidad de una hiperamoniemia primaria ante síntomas episódicos de la esfera digestiva, neurológica o psiquiátrica. El diagnóstico precoz permite instituir rápidamente el tratamiento, evitando el coma hiperamoniémico con su alta morbimortalidad.
Palabras clave: ORNITINA CARBAMOILTRANSFERASA
-deficiencia
HIPERAMONEMIA
Summary
A 2 year-old patient with neurological and recurrent digestive symptoms with laboratory analysis consistent with primary hyperammonemia by ornithine transcarbamylase deficiency is presented. Lab studies and clinical manifestations were satisfactory once treatment started. Eventhough the symptoms are nonspecific, this disease should be suspected in children with neurological, digestive or psychiatric symptoms. Early diagnosis and treatment avoids hyperammonemia coma.
Key words: ORNITHINE CARBAMOYLTRANSFERASE
-deficiency
HYPERAMMONEMIA
Introducción
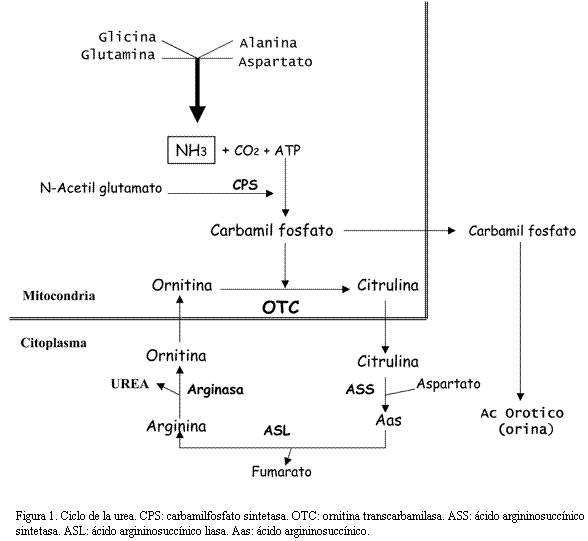
El amoníaco (A) es un producto de desecho metabólico tóxico para el sistema nervioso central que proviene de la desaminación de los aminoácidos. El ciclo de la urea (CU) en el hígado convierte el A en urea, un producto hidrosoluble, no tóxico, que se elimina por la orina (1). El CU completo se expresa sólo en el hígado con la participación de seis enzimas, una de las cuales es la ornitina transcarbamilasa (OTC) (figura 1). Se estima una frecuencia global para los defectos del CU de 1:30.000 nacimientos siendo la deficiencia de OTC probablemente la más frecuente (1-3). Su mecanismo de herencia es ligado al cromosoma X. Exceptuando pocas mutaciones recurrentes o prevalentes, la mayoría de los casos tienen mutaciones únicas o privadas (1,4,5). Una característica de la deficiencia de OTC es la gran variabilidad clínica en las mujeres, lo que podría ser una consecuencia de la proporción de hepatocitos en los cuales el cromosoma X activo porta el alelo mutado (6,7). Si bien la mayoría de los casos detectados son mujeres, han sido reportados casos en varones los cuales pueden permanecer asintomáticos incluso hasta la edad adulta (6,8,9).
Las manifestaciones clínicas se deben a la acumulación de niveles variables de A y glutamina dependiendo del grado de deficiencia enzimática (1-4). Se describe una forma de inicio neonatal grave y una forma de inicio tardío (1-4,8). Esta última forma se manifiesta en la infancia o en la adultez, es episódica y se caracteriza por vómitos, alteraciones neurológicas como ataxia, confusión mental, trastornos de conducta, manifestaciones psiquiátricas (inquietud, agresividad, alucinaciones, ansiedad), convulsiones o coma de etiología incierta. Se han consignado en algunos casos, retraso en el desarrollo y dificultad en el ascenso ponderal así como anorexia (1-3). La mujeres portadoras del alelo mutado para OTC, se encuentran en riesgo de coma hiperamoniémico en el período puerperal (1,10).
El mecanismo por el cual el A determina la encefalopatía no está totalmente esclarecido si bien se conoce que interfiere con la producción de energía y con el normal funcionamiento de los neurotransmisores (1-4).
El perfil bioquímico característico de la deficiencia de OTC es: hiperamoniemia, aumento de glutamina en sangre y aumento de ácido orótico en orina. El diagnóstico puede confirmarse evaluando la actividad enzimática en tejido hepático y/o detectando la mutación a nivel molecular (1).
Los objetivos del tratamiento son corregir las alteraciones bioquímicas y asegurar el aporte de los nutrientes necesarios. La estrategia es una reducción en la ingesta proteica, el uso de vías alternativas para la excreción de nitrógeno y el aporte de los nutrientes deficientes (11). En algunas situaciones especiales puede ser necesario el transplante hepático (12).
Estos pacientes se encuentran en permanente riesgo de descompensación aguda y grave ante los estados hipercatabólicos. El pronóstico depende del grado de deficiencia enzimática y está estrechamente relacionado con la edad del paciente y su situación clínica al momento del diagnóstico así como con la precocidad con que se inicie el tratamiento y con su cumplimiento a largo plazo (1-3).
Se presenta el caso clínico de una preescolar de dos años y tres meses con sintomatología neurológica y digestiva recurrentes, con perfil bioquímico consistente con deficiencia de OTC y evolución clínica y bioquímica satisfactoria al iniciar el tratamiento.
Caso clínico
Preescolar de dos años y tres meses, de sexo femenino, que consulta en Departamento de Emergencia del Centro Hospitalario Pereira Rossell por una convulsión tónico-clónica generalizada y breve. Es producto de segunda gestación, embarazo adecuadamente controlado y mal tolerado por hipertensión arterial. Se realizó cesárea a término. Peso al nacimiento 3.850 gramos, talla 47 cm. Desconocemos el puntaje de Apgar y el perímetro craneano al nacer. Se otorgó el alta sin enfermedad perinatal. Presentó adecuado desarrollo. Fue alimentada con pecho directo exclusivo tres meses y recibió pecho hasta los dos años de vida. Al momento de la consulta recibía dieta lacto-vegetariana y presentaba rechazo por la carne. Destacamos que presentó al año de vida un episodio caracterizado por alternancia de excitación-depresión y vómitos de 24 horas de evolución tratado con hidratación intravenosa. Posteriormente consultó múltiples veces por vómitos. En reiteradas oportunidades presentó episodios de inestabilidad en la marcha y temblores finos distales que cedieron espontáneamente. Desde el inicio de la sintomatología muestra trastornos de la conducta caracterizados por insomnio, inquietud e irritabilidad que alteran seriamente la vida familiar.
Al examen se evidencia hiperquinesia e irritabilidad, siendo el resto del examen físico normal. Peso 11.300 g Talla: 87 cm. Perímetro cefálico: 49 cm.
De los exámenes complementarios destacamos:
- Amoniemia post ingesta proteica: 3.17 µg /ml (valor normal: 0,20-1,04 µg /ml).
- Acido orótico urinario post ingesta proteica (valor normal: hasta 10 mg/g creatinina):
- Muestra Nº 1: 498 mg/g creatinina.
- Muestra Nº 2: 378 mg/g creatinina.
- Aminoácidos plasmáticos (en hiperamoniemia).
- Glutamina: 1.192 µmol/l (valor normal: 254-823 µmol/l).
- Citrulina: 11 µmol/l (valor normal: 1-46 µmol/l).
- Ornitina: 51 µmol/l (valor normal: 48-284 µmol/l).
- Arginina: 56 µmol/l (valor normal 10-140 µmol/l).
- Electroencefalograma: discreta actividad epileptógena izquierda.
Dado el perfil bioquímico consistente con deficiencia de OTC, se inició el tratamiento limitando la ingesta proteica y asegurando un buen aporte calórico. Al inicio de la dieta, hubo muy escaso ascenso ponderal por lo cual se aumentó la cantidad de proteínas de la dieta y se adicionó benzoato de sodio.
La evolución ha sido favorable, con mejoría clínica del carácter, la inquietud y del trastorno del sueño. Su desarrollo neuropsíquico se ha mantenido dentro de lo normal. No ha reiterado episodios de vómitos ni sintomatología neurológica. Ha presentado un ascenso pondoestatural con curvas de crecimiento de peso y estatura en el percentil 5. En el seguimiento bioquímico, los valores de oróticoaciduria y amoniemia posprandial han permanecido dentro de los rangos normales.
Comentarios
Aproximadamente la mitad de los casos de los errores congénitos del metabolismo intermediario (ECMI) de inicio tardío (más allá de los primeros meses de vida) se presentan con una expresión clínica aguda intermitente (13). El período libre de síntomas puede extenderse incluso hasta la edad adulta. Los síntomas agudos y recurrentes que pueden inaugurar un ECMI son variados. Quizás los más comunes sean: vómitos, ataxia, síntomas psiquiátricos, coma. Se trata de episodios de intoxicación endógena determinados muchas veces por un estado catabólico, como fiebre o ayuno, pero a veces sin una causa clara. La evolución del episodio puede ser hacia la resolución rápida sin secuelas, con secuelas neurológicas o inexorablemente a la muerte. Es por esto muy importante intentar establecer el diagnóstico lo antes posible (14).
En el caso que se presenta, si bien la paciente ingresó por una crisis convulsiva generalizada, no fue lo que orientó el diagnóstico. Sus antecedentes de vómitos reiterados, un episodio de excitación–depresión sin causa clara al año de vida, los trastornos de conducta con irritabilidad e hiperquinesia y algún episodio de inestabilidad en la marcha recordaron la presentación de tipo intoxicación de los errores congénitos del metabolismo del CU. Estudiada con esa orientación, el perfil bioquímico fue consistente dentro de las hiperamoniemias primarias, con una deficiencia de OTC: hiperamoniemia, glutamina plasmática elevada, ácido orótico urinario aumentado con citrulina, ornitina y arginina normales. Ninguna de las otras deficiencias enzimáticas del CU podría explicar este perfil bioquímico. Al igual que en algunas otras publicaciones (7,8), el criterio diagnóstico de deficiencia de OTC para el caso presentado, está basado en el perfil bioquímico.
En un trabajo que revisa la historia natural de la deficiencia de OTC en 13 casos de pacientes de sexo femenino, con edades de inicio entre una semana y seis años, el porcentaje de síntomas es el siguiente: episodios de irritabilidad extrema 100%; episodios de vómitos y letargia 100%; rechazo de proteínas 92%; ataxia 77%; coma estadio II 46%; retraso del crecimiento 38%; retraso del desarrollo 38%; y convulsiones 23%. El intervalo medio entre el inicio de los síntomas mayores y el diagnóstico fue de 16 meses (7). El caso que se presenta tiene historia de irritabilidad, vómitos episódicos, rechazo de proteínas e inestabilidad en la marcha en alguna oportunidad, elementos encontrados en más del 90% de los casos del referido trabajo.
Las pacientes con deficiencia de OTC se encuentran en permanente riesgo de una descompensación aguda y grave ante situaciones de estrés con altos requerimientos energéticos, pudiendo llegar al coma hiperamoniémico que tiene una elevada morbimortalidad, probablemente por edema cerebral (1,2). El paciente en coma que sobrevive generalmente queda con secuelas neurológicas irreversibles. Es por ello muy importante mantener un alto grado de alerta por parte de la familia y del pediatra para iniciar rápidamente el tratamiento de las descompensaciones ante los primeros síntomas (2,15). El tratamiento crónico debe ser controlado regularmente, tanto clínica como bioquímicamente, a fin de realizar los ajustes necesarios para evitar aumento de A plasmático y deficiencia de aminoácidos esenciales (16,17). En el caso que presentamos la adaptación a la dieta fue fácil, dado que la paciente tenía rechazo por los alimentos ricos en proteínas. En los primeros meses de iniciado el tratamiento el ascenso ponderal fue escaso, por lo que se introdujo benzoato de sodio para aumentar la eliminación de nitrógeno y poder aumentar la cuota proteica diaria sin que aumente el amonio postprandial. Con esta medida se mejoró la incorporación de peso, si bien permaneció en el percentilo 5. Por otro lado, luego del inicio de la dieta, se notó por parte de los padres un favorable cambio en el comportamiento y en el sueño nocturno. Algunos pacientes presentan anorexia, lo que puede hacer difícil su adaptación a la dieta. Medicamentos como ciproheptadina pueden mejorar el apetito en alguno de estos casos, lo que facilita su manejo nutricional (18). Si bien en esta deficiencia enzimática del CU el transplante hepático es excepcional, en algunos casos puede ser necesario (12).
Una vez que el diagnóstico de deficiencia de OTC ha sido establecido en una familia, es necesario investigar la historia familiar detalladamente y realizar búsqueda de posibles mujeres portadoras. Si se ha identificado la mutación, sería ésta la mejor forma para su investigación (6,9). De no contar con esta información, lo más conveniente para algunos autores es el test del alopurinol, que parecería tener alta sensibilidad y especificidad (15). Para otros autores, es un método que podría tener errores (19,20). Las portadoras sintomáticas deben iniciar tratamiento. En todos los casos las portadoras (sintomáticas y no sintomáticas) deben ser advertidas de una eventual descompensación en el posparto inmediato (1,10). Se estima que 15% de las mujeres portadoras presentan hiperamoniemia en algún momento de su vida (21). En la familia que se presenta, no ha sido posible comenzar aún con la búsqueda de portadoras.
En la bibliografía revisada a nivel nacional, no se encontraron publicaciones sobre hiperamoniemia primaria por deficiencia de OTC, sería ésta la primera publicación.
ConclusionesSi bien los síntomas aislados son totalmente inespecíficos, es nuestro interés subrayar con este trabajo la importancia de mantener la sospecha de las hiperamoniemias primarias ante síntomas episódicos de la esfera digestiva, neurológica o psiquiátrica. El diagnóstico precoz permitirá instituir rápidamente el tratamiento evitando el coma hiperamoniémico y su alta morbimortalidad. Además, el diagnóstico permite el asesoramiento genético a la familia.
Bibliografía
1. Brusilow SW, Horwich AL. Urea cycle enzymes. In: Scriver Ch R, Baudet AL, Sly WS, Valle MD, editores. Eight edition. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw Hill, 2001: 1187-232.
2. Sanjurjo P, Rubio V. Diagnóstico y tratamiento de las enfermedades del ciclo de la urea. In: Sanjurjo P, Baldellou A, editores. Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. Madrid: Ergon, 2001: 324-32.
3. Rezvani I. Defects in Metabolism of Aminoacids. In: Behrman RE, Kliegman RM, Jenson HB Editors. Nelson Textbook of Pediatrics. 16th Edition. Philadelphia: WB Sanders, 2000: 344–76.
4. Tuchman M. The clinical, biochemical, and molecular spectrum of onithine transcarbamylase deficiency. J Lab Clin Med 1992; 120(6): 836-50.
5. Tuchman M, McCullough B, Yodkoff M. The molecular basis of ornitine transcarbamilase deficiency. Eur J Pediatr 2000; 159: S196-S198.
6. Ausems M, Bakker E, Berger R, Duran M, Van Diggelan O, Keulemans J, et al. Asymptomatic and late - onset ornithine transcarbamylase deficiency caused by a a208t mutation: clinical, biochemical, and dna analyses in a four - generation family. Am J Med Genet 1997; 68: 236-9.
7. Rowe PC, Newman SL, Brusilow SW. Natural history of symptomatic partial ornithine transcarbamylase deficiency. N Engl J Med 1986; 314(9): 541-7.
8. Matsuda I, Nagata N, Matsuura T,Oyanagi K, Tada K, Narisawa K, et al. Retrospective Survey of Urea Cycle Disorders: Part 1: Clinical and Laboratory Observations of Thirty-Two Japanese Male Patients With Ornithine Transcarbamylase Deficiency. Am Hum Genet 1991; 38: 85-9.
9. McCullough B, Yodkoff M, Batshow M, Wilson J, Roper S, Tuchman M. Genotype Spectrum of Ornithine Transcarbamylase Deficiency. Correlation with the clinical and Biochimical Phenotype. Am J Med Gen 1999; 93: 268-72.
10. Hawks P, Hauser ER,Thomas GH, Herman G, Hess D, Brusilow SW. Brief Report. Hiperammoniemia in women with a mutation at the Ornithine Carbamoyltransferase locus . A Cause of Postpartum Coma. N Engl J Med 1990; 332(23): 1652-6.
11. Maestri N, Hauses E, Bartholomew D, Brusilow S. Prospective Treatment of Urea Cycle Disorders. J Pediatr 1991; 119: 923-8.
12. Saudubray J, Touati G, Delonlay P, Jouvent P, Narcy C, Laurent D, et al. Live transplantation in Urea Cycle disorders. Eur J Pediatr 1999; 158: 55-9.
13. Saudubray JM, Ogier de Baulny H, Charpentier C. Clinical approach to inherited metabolic diseases. In: Fernandes J, Saudubray JM, Van den Berghe G, eds. Inborn Metabolic Diseases. Diagnosis and Treatment. Berlin: Springer-Verlag, 2000: 4-41.
14. Chamoles NA. Urgencias Metabólicas y Errores Congénitos del Metabolismo. En: Fejerman, Fernández Alvarez, editores. Neurología Pediátrica. Segunda edición. Buenos Aires: Médica Panamericana, 1997: 301-19.
15. Leonard V. Disorders of the Urea Cycle. In: Fernandes J, Saudubray JM, Van den Berghe G, eds. Inborn Metabolic Diseases. Diagnosis and Treatment. Berlin: Springer-Verlag, 2000: 215-22.
16. Berry G, Steinert R. Long-term management of patients with urea cycle disorders. J Pediatr 2000; 138: S56-S61.
17. Wilson CJ, Lee PJ, Leonard JV. Plasma glutamine and ammonia concentrations in ornithine carbamoyltransferase deficiency and citrullinaemia. J Inherit Metab Dis 2001: 24: 691-5.
18. Lerman-Sagie T, Minouni M. Reversal of Anorexia in a Child With Partial Ornithine Transcarbamilase Deficiency by Cyproheptadine Therapy. Clin Pediatr 1995; 163-5.
19. Barshop BA, Nyham WL, Climent C, Rubio V. Short report. Pitfall in the detection of heterozygosity by allopurinol in a variant form of ornithine carbamoyltransferase deficiency. J Inherit Metab Dis 2001; 24: 573-4.
20. Potter M, Hammond JW, Sim KG, Green AK, Wilken B. Ornithine carbamoyltransferase deficiency: improved sensitivity of testing for protein tolerance in the diagnosis of heterozygotes. J Inherit Metab Dis 2001; 24: 5-14.
21. Brusilow S. Urea cycle disorders: clinical paradigm of hyperammonemic encephalopathy. Prog Liver Dis 1995;13: 293-309.
Correspondencia: Dra. Aída Lemes.
E-mail: lemesa@adinet.com.uy


{kind=link}