











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Introducción
En Génova, en 1998, en la segunda reunión de consulta sobre la hipercolesterolemia familiar (HF) organizada por la Organización Mundial de la Salud (OMS), se generó un reporte de expertos de 33 países que afirmaba en su primer párrafo: “Se estima que cerca de 250 millones de personas en el mundo están expuestas a riesgo muy alto de muerte a una joven edad porque son portadoras de uno o más genes que promueven desórdenes lipídicos heredables. Estos incluyen la hipercolesterolemia familiar (estimada en 10 millones), la hipercolesterolemia familiar combinada (HFC, 40 millones) y la hipercolesterolemia poligénica severa (HP, estimada en 200 millones)”1.
Aunque esta afirmación sigue siendo válida, la advertencia sobre las consecuencias en la salud de los adultos jóvenes no fue tenida en cuenta en primera instancia. La Sociedad Europea de Aterosclerosis, 15 años después, publicó un consenso sobre HF que mostraba el subdiagnóstico y subtratamiento en la población general y estableció guías para prevenir la enfermedad cardíaca2.
Se concluyó que frente a la severidad de la situación, era necesario establecer estrategias de diagnóstico y tratamiento precoz a nivel mundial para una condición reconocida como generadora de una alta morbilidad y mortalidad en el adulto joven, fácil de prevenir y combatir.
En el mismo documento se destacaron los instrumentos más importantes para la identificación de pacientes: 1) la identificación familiar en cascada y los screenings oportunistas; 2) la implementación del diagnóstico molecular establecido como el gold standard, explorando los genes LDLR y APOB, para superar zonas grises del diagnóstico guiado por los niveles plasmáticos de colesterol; 3) el establecimiento de registros centralizados que permitieran trabajar en el seguimiento de los pacientes y las familias generando una ayuda efectiva al médico en prevención primaria, tal como planteara el docu mento de la OMS.
La genética en la estimación del riesgo
La estimación de la susceptibilidad genética de un individuo a la enfermedad y la predicción del riesgo son fundamentales para la detección temprana y la prevención, especialmente en enfermedades que se expresan en el adulto.
Un patrón familiar de riesgo de enfermedad coronaria (EC) se describió por primera vez en 19383. El componente genético es sin duda constitutivo, está presente en el sujeto desde la concepción y con frecuencia podemos encontrar su rastro en la transmisión familiar. Una mayor concordancia de eventos clínicos entre familiares con un parentesco biológico cercano presume un mayor número de genes compartidos y mayor probabilidad de semejanza en la presentación y evolución clínica de la enfermedad.
El análisis de la historia de salud y enfermedad familiar se estableció como un instrumento valioso para el asesoramiento y la prevención del riesgo coronario4,5.
La historia familiar y su representación gráfica como genealogía médica es una valiosa fuente de datos para establecer el diagnóstico y tratamiento del caso índice y del grupo familiar.
En la HF la severidad de la presentación clínica en la familia, determinada por los niveles de colesterol unido a las lipoproteínas de baja densidad (C-LDL) en el perfil lipídico y por eventos coronarios prematuros, son los conductores más importantes del diagnóstico presuntivo y transforman la his toria familiar en el primer estimador del riesgo del paciente.
Este instrumento clásico y subutilizado de la anamnesis se ha visto enriquecido por nuevos conocimientos de las correlaciones clínico-moleculares. Sin embargo, la historia familiar (como una primera aproximación) y la identificación de variantes génicas compartidas por individuos emparentados comienzan a utilizarse con más frecuencia en las últimas décadas debido principalmente a la reducción de costos de la secuenciación6.
Las mutaciones son fuertes marcadores del riesgo coronario
La importancia de la genética en la estimación del riesgo coronario de la HF se determinó con claridad en un trabajo reciente de Khera y colaboradores, que demuestra que dentro de cualquier estrato de C-LDL observado, el riesgo de EC es mayor entre los portadores de mutaciones causantes de HF7. En individuos con C-LDL >190 mg/dl encontrar una mutación patogénica en el gen LDLR aumenta el riesgo de EC unas 22 veces (OR: 22,3; IC 95%: 10,7-53,2) (figura 1)7.
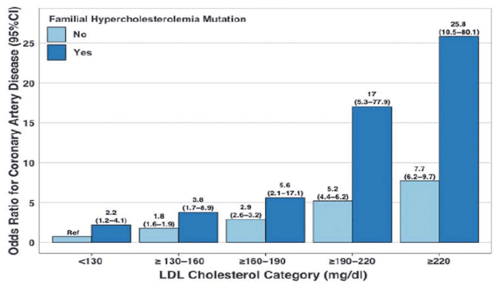
Figura 1: Impacto de las mutaciones patogénicas en LDLR en el riesgo de enfermedad coronaria. Para un mismo nivel de C-LDL una variante patogénica en LDLR aumenta significativamente el riesgo de enfermedad coronaria. Tomado de Khera y colaboradores7.
Una prevalencia más alta que la estimada
Estudios recientes de los programas de HF en la población del norte de Europa revelaron cifras de prevalencia en el orden de 1 en 200, duplicando la estimación clásica de 1 en 5009. Un estudio de la base de datos de lípidos de Estados Unidos (n >1,3 millones) sugiere que la prevalencia de HF es de 1 en 300, utilizando los criterios de la Asociación Nacional de Lípidos de Estados Unidos (National Lipid Association, NLA)10. Un cambio de esta magnitud modifica las cifras esperadas de HF en el mundo de 14 a 34 mi llones.
La prevalencia de la HF homocigota (clásicamente considerada muy baja) en el orden de 1 en 1.000.000, debe esperarse ahora en frecuencias entre 1 en 160.000-300.000 individuos11.
Estas cifras, que se están confirmando en todo el mundo, revelan un importante retraso diagnóstico y la existencia de niños y jóvenes afectados sin atención.
En Latinoamérica la situación es similar en cuanto a prevalencia, subdiagnóstico y subtratamiento según un trabajo reciente de la Red Iberoamericana de HF, que incluyó datos de Argentina, Brasil, Chile, México, Uruguay, España y Portugal12).
En Uruguay, la prevalencia todavía no es conocida. Una aproximación práctica basada en análisis de perfiles lipídicos hospitalarios demuestra una fre cuencia en el orden de 1 en 300 cuando se seleccionan niveles de C-LDL > 250 mg/dl con triglicéridos normales (datos del Registro Nacional de HF, aún no publicados).
Considerando la prevalencia estimada por este método, en nuestro país deberíamos encontrar aproximadamente 12.000 portadores de HF heterocigota y 11 homocigotos.
Estos pacientes constituyen una población altamente vulnerable que exige la implementación de políticas específicas tendientes a disminuir la morbilidad y mortalidad por esta condición.
Una proyección a futuro, considerando que en Uruguay se producen 42.000 nacimientos anuales, sugiere que nacerían 140 niños portadores de HF por año, con una acumulación de 4.200 casos cada 30 años, en base a una estimación conservadora de un caso cada 300 nacimientos.
Los genes y las variantes causales
La HF es un trastorno heterogéneo del metabolismo lipídico determinado por variantes patogénicas raras (frecuencia alélica menor a 1%) de efecto mayor en genes involucrados en el metabolismo del colesterol, y por variantes comunes con poco efecto en forma individual, pero que en conjunto determinan una HP13,14. Estos mecanismos interaccionan entre sí (herencia monogénica y herencia poligénica) y con el ambiente para determinar el amplio espectro de fenotipos clínicos en los pacientes con HF.
La cantidad de genes afectados también tiene influencia en el fenotipo. Enumeramos los genes más importantes:
1.LDLR. Es el gen que codifica el receptor de LDL (RLDL). El RLDL reside en la membrana. plasmática y es responsable de la absorción de partículas de C-LDL hacia la célula, donde son degradadas15. El 90% de las mutaciones que determinan una HF se encuentran en este gen y se transmiten de forma autosómica dominante16. Por lo tanto, la enfermedad se ma nifiesta en heterocigotos con un alelo mutado de LDLR o en pacientes con dos alelos mutados que se denominan heterocigotos compuestos (cuando son dos variantes distintas), u homocigotos cuando presentan la misma alteración en los dos alelos del gen. Debido a esto, en heterocigotos compuestos, pero sobre todo en homocigotos, el hecho de que no exista una forma normal del gen determina un fenotipo más severo17. Se han descripto más de 2.900 variantes en este gen que afectan todos los dominios funcionales y por lo tanto todos los estadios del ciclo intracelular del RLDL18. La función de este gen se ve afectada por distintos tipos de mutaciones: rearreglos genéticos (deleciones o inserciones grandes), mutaciones sin sentido (stop prematuro), mutaciones de cambio de sentido, mutaciones que afectan los sitios de splicing y mutaciones en el promotor que disminuyen la expresión del gen18. Este amplio espectro de mutaciones puede requerir de más de una técnica de biología molecular para identificarlas. Debido a que las manifestaciones clínicas en el heterocigoto en general aparecen luego de la edad reproductiva, la expansión de la mutación hacia la descendencia es la regla.
2.APOB. Este gen codifica la proteína por la que el RLDL se une a las partículas de C-LDL y promueve su captación. Las variantes patogénicas en este gen llevan también a un aumento de la concentración plasmática de C-LDL y EC prematura19. Las mutaciones en APOB más frecuentes se ubican dentro de la secuencia que codifica el dominio de unión al RLDL, y causan un tipo de HF que también se conoce como apo B defectuosa familiar (FDB) con fenotipo menos grave20. Entre 5% y 10% de los pacientes con diagnóstico clínico de HF tienen mutaciones en este gen. Son más comunes en Europa central y en poblaciones de origen celta y gallegas, donde la prevalencia de FDB es mayor que en otras áreas21.
3.PCSK9. Codifica una proteína (proproteína convertasa subtilisina/kexina 9) que actúa como un antagonista del RLDL al promover su degradación. Las pocas mutaciones conocidas de ganancia de función (< 1% en todas las series) causan hipercolesterolemia al inhibir el clearance de C-LDL, y una HF dominante semejante a las del gen LDLR22. Las mutaciones de pérdida de función son más frecuentes y causan una disminución constitutiva del nivel de C-LDL y del riesgo de EC. Esta proteína se ha transformado en un objetivo terapéutico exitoso, ya que su inhibición promueve el aumento de RLDL y una disminución de los niveles de C-LDL por un mecanismo diferente a las estatinas o captación de colesterol, por lo que se suman al arsenal terapéutico en pacientes con resistencia o mala respuesta a los tratamientos clásicos23.
4.LDLRAP1. La genética de la hipercolesterolemia autosómica recesiva (ARH) se asocia a mutaciones en el gen que codifica la proteína adaptadora del RLDL 1 (LDLRAP1), proteína citosólica que contiene un dominio de unión a fosfotirosina, que se une directamente a la cola citoplásmica del receptor y media su internalización celular por el RLDL de clatrinas24. Las mutaciones en este gen conducen al mal funcionamiento del RLDL y causan la enfermedad25.
5.ABCG5 y ABCG8. Estos genes codifican para dos proteínas esterolina -1 y -2, que son esenciales para la regulación de la absorción y excreción de esteroles. Las mutaciones en cualquiera de ellos provocan un trastorno lipídico, llamado sitosterolemia. Esta enfermedad de herencia autosómica recesiva es poco frecuente y genera una fenocopia de la HF, con altos niveles de esteroles en plasma y aumento de C-LDL. Los hallazgos clínicos incluyen xantomas, artralgias y aterosclerosis prematura26.
La HF es una población de alto nivel de riesgo de EC de comienzo temprano, con una vulnerabilidad genética identificable causada principalmente por mutaciones en los genes LDLR, APOB y PCSK98. Si bien prácticamente el 95% de las mutaciones se encuentran en estos genes, existen casos muy raros con mutaciones en genes como LIPA, STAP1 y APOE27-29) (tabla 1).
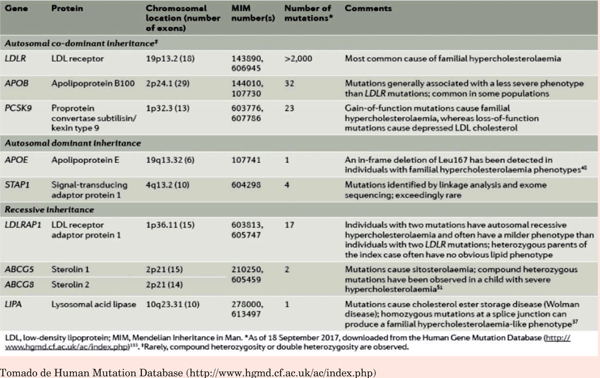
Con los avances de la secuenciación masiva los genes mencionados pueden ser secuenciados a un costo razonable para identificar las mutaciones causantes de HF13. Estas tecnologías han tenido un gran impacto en el diagnóstico genético.
Sin embargo, uno de los principales problemas derivados del uso de estas tecnologías es el descubrimiento de un gran número de variantes de significado incierto (VUS, del inglés Variations of Unknown Significance). En particular, aparecen con una alta frecuencia en el gen APOB, un gen muy largo y hasta ahora poco explorado en el que se han encontrado mutaciones que aumentan o disminuyen los niveles de colesterol circulante20,30. Las bases de datos internacionales que comparten información genética, junto con la clínica, son importantes para avanzar en la determinación de la patogenicidad de estas variantes.
El aporte de los polimorfismos simples: los scores de riesgo genético
El desarrollo de estudios de asociación de genoma completo (GWAS, del inglés Genome Wide Association Studies) permitió identificar numerosas variantes comunes de alta frecuencia (frecuencia alélica mayor a 5%) asociadas a niveles de colesterol y a riesgo de EC8,14.
El conocimiento de estas variantes fue clave para el desarrollo de scores poligénicos que pueden explicar niveles elevados de C-LDL en plasma en ausencia de mutaciones mayores en genes clásicos31,32. Esta forma de HF, conocida como HP, está presente hasta en el 80% de los pacientes con HF sin mutaciones de efecto mayor14.
El score poligénico puede ser de utilidad en la practica clínica. Permite establecer un diagnóstico de causa genética en pacientes sin mutaciones en genes conocidos, e identificar algunos pacientes que podrían beneficiarse de un tratamiento menos agresivo14).
Dislipemias aterogénicas y enfermedad coronaria precoz
Estudios recientes han intentado determinar la susceptibilidad genética a la EC prematura mediante la estrategia de secuenciación del exoma completo. Mediante esta estrategia, Do y colaboradores identificaron variantes patogénicas poco frecuentes en pacientes con infarto de miocardio precoz (menores de 50 años en los varones y 60 años en las mujeres) en los genes LDLR y APOA5, vinculados al metabolismo del C-LDL y triglicéridos, respectivamente33. A su vez, un trabajo reciente de Khera y colaboradores demuestra que variantes en el gen LPL están asociadas a niveles elevados de triglicéridos y a mayor riesgo de infarto agudo de miocardio34. Estas observaciones reafirman el rol del C-LDL como factor de riesgo para EC y posicionan a la hipertrigli ceridemia como un gran factor de riesgo para el de sarrollo de esta enfermedad.
Menos eventos con niveles de C-LDL bajo desde el nacimiento
Michael S. Brown y Joseph L. Goldstein, los descubridores del RLDL que recibieron por este trabajo el Premio Nobel de Medicina en el año 1986, publicaron un comentario en la revista Science35, que titularon “Reducir el C-LDL: no solo cuán bajo, sino ¿cuánto tiempo?”. El comentario refiere al estudio de Cohen y colaboradores36, que analizando a estadounidenses de mediana edad, encontraron un pequeño número de sujetos con mutaciones nulas en PCSK9, en quienes la concentración de C-LDL se redujo en 38 mg/dl, pero la prevalencia de EC disminuyó 88%. En estas familias los niveles extremadamente bajos de C-LDL desde el nacimiento son naturalmente protectores de la aterosclerosis coro naria y disminuyen la EC más allá de lo que se logra con los hipolipemiantes disponibles.
Otros trabajos, como los de Ference y colaboradores37, reafirmaron que la exposición prolongada a niveles más bajos de colesterol disminuye el riesgo de EC.
La reducción del C-LDL de comienzo temprano en la vida puede prevenir o retrasar sustancialmente la progresión de la aterosclerosis coronaria y, por lo tanto, mejorar significativamente el beneficio clínico de las terapias hipolipemiantes.
Conclusiones
La HF es un problema de salud pública en todo el mundo. Distintos programas internacionales han promovido un “llamado a la acción”, tanto a gobiernos nacionales como a médicos, para disminuir la morbilidad y mortalidad por esta enfermedad38,39. Los avances en los conocimientos de las bases genéticas de la HF impactan fuertemente en el diagnóstico, tratamiento y control de esta enfer medad.
El diagnóstico de HF se confirma al encontrar una variante patogénica en alguno de los genes causantes. Una mutación patogénica en un paciente con sospecha clínica de HF aumenta significativamente el riesgo de EC y en algunos casos justifica un tratamiento de mayor intensidad para alcanzar los objetivos terapéuticos. A su vez, la identificación de una variante patogénica en el caso índice permite iniciar la identificación familiar en cascada a un bajo costo. Esta estrategia ha demostrado ser la más costo-efectiva, ya que permite detectar pacientes en su etapa presintomática y antes del desarrollo de aterosclerosis significativa. La detección precoz de la HF permite implementar tratamientos para reducir el C-LDL temprano en la vida de los pa cientes, mejorando los resultados clínicos.
En pacientes con sospecha clínica de HF y sin mutaciones en genes clásicos, el uso de scores poligénicos ayuda a definir la etiología de los altos niveles de colesterol. Este grupo de pacientes con un score positivo tienen un menor riesgo cardiovascular que las HF monogénicas, y en algunos casos puede estar justificado un tratamiento de menor intensidad.
De esta forma, el diagnóstico genético se ha posicionado como una herramienta de prevención primaria con potencial para establecer con mayor precisión el riesgo cardiovascular y personalizar el tratamiento en aquellos con mayor vulnerabilidad genética.

