











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo

 Permalink
Permalink
Introducción
El HC es una enfermedad rara que resulta de la deficiencia de una o más hormonas hipofisarias desde el nacimiento. Puede presentarse como un déficit aislado, principalmente de hormona de crecimiento (GH), o como una deficiencia combinada de múltiples ejes, afectando en distintos grados al eje somatotropo, tirotropo, corticotropo, gonadotropo e incluso al eje arginina-vasopresina1,2.
La incidencia estimada varía entre 1 por cada 4.000 y 1 cada 16.000 nacidos vivos, dependiendo de las series y los criterios diagnósticos utilizados3–5. En estudios europeos, la mayoría de los casos se detectan en el período neonatal, aunque el diagnóstico puede retrasarse si la presentación clínica es leve o inespecífica6,7.
Las manifestaciones clínicas varían según el número y tipo de ejes hormonales comprometidos. En la etapa neonatal, los signos más comunes incluyen hipoglucemia, ictericia, micropene y criptorquidia, siendo estas dos últimas exclusivas del sexo masculino8–10. En casos de diagnóstico tardío, el motivo de consulta suele ser talla baja o retraso puberal11,12.
La resonancia magnética (RM) es una herramienta esencial para la evaluación estructural del eje hipotálamo-hipofisario. Entre las anomalías más frecuentes se encuentran la hipófisis hipoplásica o ausente, la neurohipófisis ectópica y la interrupción del tallo hipofisario, patrón que define el síndrome de interrupción del tallo hipofisario13–15.
Si bien se han identificado mutaciones en genes como PROP1, POU1F1, GH1, HESX1 y LHX3, la causa genética del HC se confirma solo en un porcentaje menor de casos16–18. En muchos países, como Uruguay, el acceso limitado a estudios moleculares constituye una barrera diagnóstica.
La pesquisa neonatal, basada en la medición de TSH en sangre de cordón o gota seca, permite detectar el hipotiroidismo congénito primario. Sin embargo, no permite identificar los casos de hipotiroidismo central, lo que genera un subregistro de pacientes con HC19–21.
En Uruguay, no se dispone de estudios publicados que caractericen clínicamente esta enfermedad en la población pediátrica. Por lo tanto, el objetivo de este trabajo es describir las características clínicas, hormonales, imagenológicas y terapéuticas de los pacientes con HC atendidos en el centro de referencia del país durante el período 2012–2022.
Materiales y métodos
Diseño del estudio
Se realizó un estudio observacional, descriptivo y retrospectivo. Se revisaron historias clínicas de pacientes con diagnóstico de hipopituitarismo congénito en seguimiento entre enero de 2012 y diciembre de 2022.
Población de estudio
Se incluyeron pacientes entre 0 y 21 años, con diagnóstico confirmado de deficiencia congénita de una o más hormonas hipofisarias, en tratamiento hormonal sustitutivo, activo o previo, en la policlínica de Endocrinología del Hospital Pereira Rossell.
Variables
Se analizaron las siguientes variables:
Clínicas: edad al diagnóstico, síntomas iniciales.
Bioquímicas: perfil hormonal (GH,IGF-1, TSH, T4L, ACTH, cortisol de hora 8, FSH, LH, PRL, ADH).
Imagenológicas: hallazgos en resonancia magnética de cráneo.
Terapéuticas: tipo de tratamiento sustitutivo recibido.
Epidemiológicas: incidencia estimada en el período.
Métodos diagnósticos
La deficiencia hormonal se diagnosticó mediante evaluación clínica y análisis hormonales séricos, considerando rangos de referencia por edad. La resonancia magnética se utilizó para estudiar la anatomía del eje hipotálamo-hipofisario. En ninguno de los pacientes se realizaron estudios genéticos.
Evaluación clínica
Se relevaron retrospectivamente los signos clínicos documentados en la historia clínica. Las variables clínicas consideradas fueron las siguientes:
Hipoglucemia: glucemia venosa <40 mg/dL (según pauta del servicio de neonatología del CHPR).
Micropene: consignado como longitud del pene menor a −2 desvíos estándar (DE) de la media para la edad gestacional.
Criptorquidia: ausencia documentada de uno o ambos testículos en bolsa escrotal al nacimiento.
Talla baja: valor de estatura al momento del diagnóstico menor a −2 DE para la edad y sexo.
Ictericia: presencia consignada en la historia clínica de coloración amarilla de piel y mucosas en el período neonatal.
Normas éticas
El estudio fue aprobado por el Comité de Ética del Hospital Pereira Rossell. Se garantizó la confidencialidad y anonimato de los datos, de acuerdo con las normativas nacionales de protección de datos personales (Ley 18331).
Análisis estadístico
Se utilizó estadística descriptiva para las variables cuantitativas (media, mediana, desviación estándar) y cualitativas (frecuencias absolutas y relativas). Para el análisis de asociaciones entre variables categóricas se empleó la prueba exacta de Fisher. Se consideró significativo un valor de p < 0,05. El análisis fue realizado con el programa IBM® SPSS®.
Resultados
Se identificaron 12 pacientes con diagnóstico de hipopituitarismo congénito en el período 2012–2022. La distribución por sexo fue equitativa (50% masculino y 50% femenino). Nueve pacientes (75%) fueron diagnosticados antes de los seis meses de vida y tres (25%) en la etapa escolar (Tabla 1).
Tabla 1. Características clínicas, hormonales e imagenológicas de los 12 pacientes con diagnóstico de hipopituitarismo congénito entre 2012 y 2022.
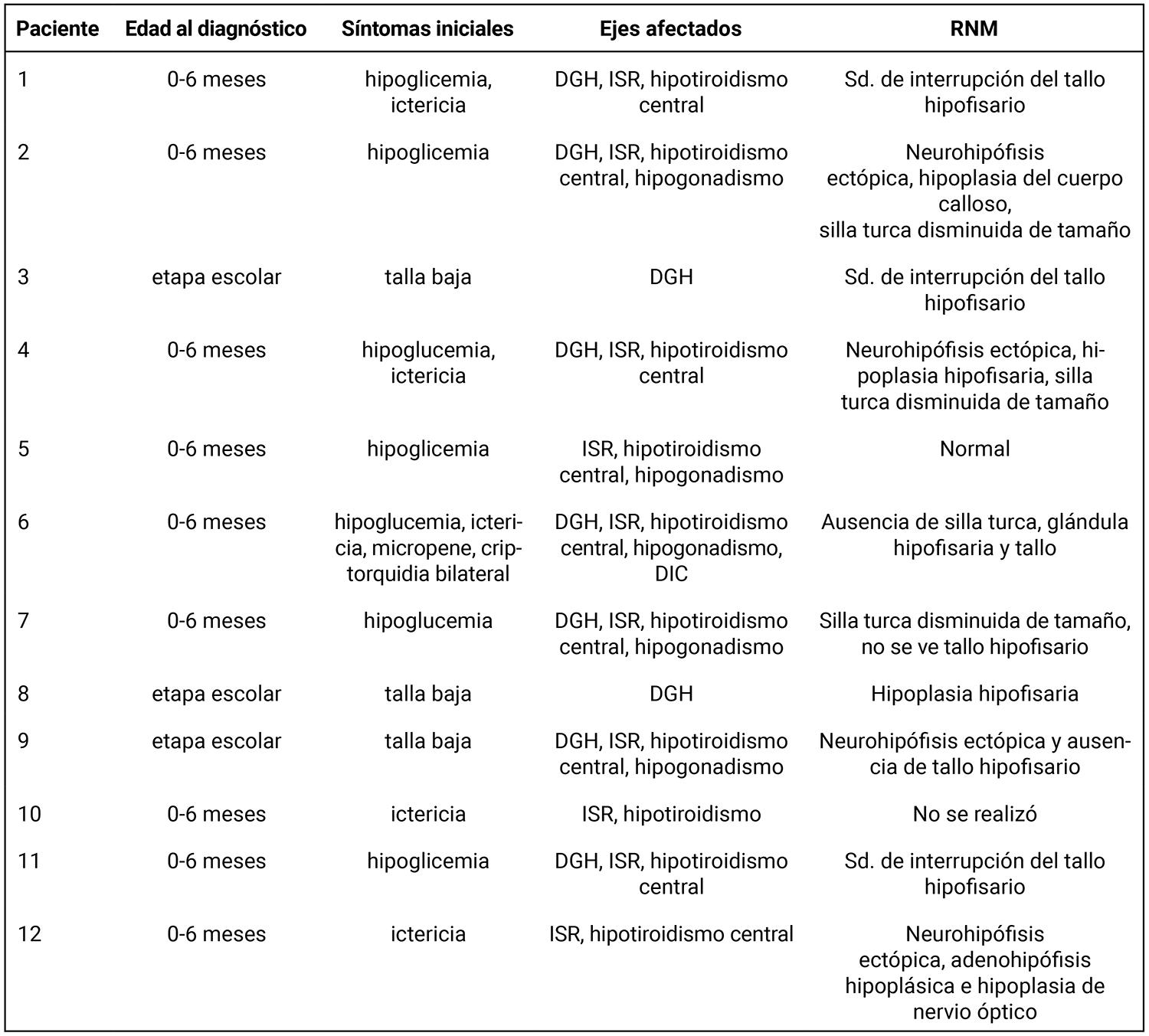
DGH: déficit de hormona de crecimiento; ISR: insuficiencia suprarrenal; DIC: diabetes insípida central; RNM: resonancia nuclear magnética.
Síntomas iniciales y edad al diagnóstico
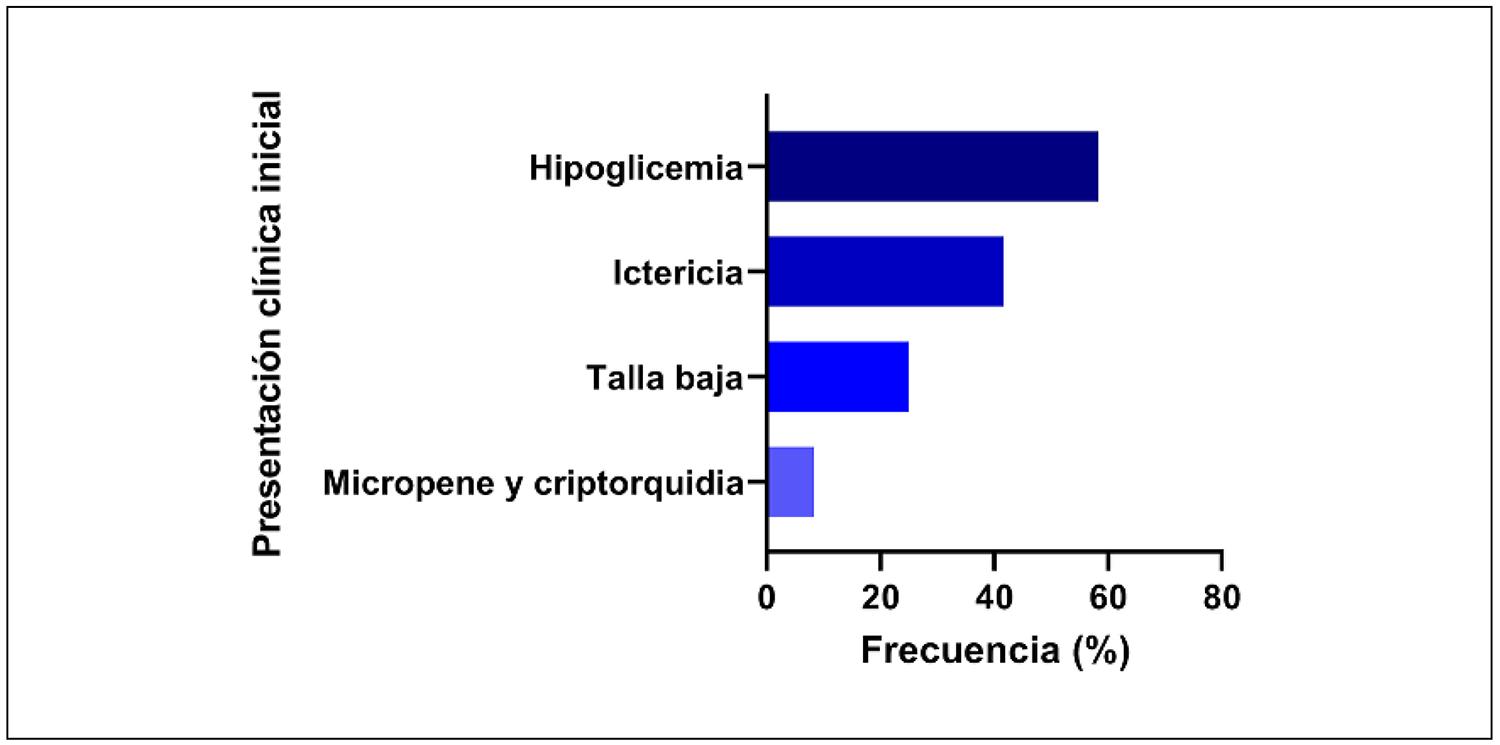
En los pacientes diagnosticados antes de los seis meses (nueve) los síntomas iniciales más frecuentes fueron: hipoglucemia en siete casos (77,7%), ictericia en cinco (55,6%) y micropene/criptorquidia en uno (11,1%). Los tres pacientes diagnosticados en etapa escolar presentaron talla baja como único motivo de consulta. Se encontró una asociación estadísticamente significativa entre hipoglucemia y edad temprana al diagnóstico (p = 0,045), así como entre talla baja y diagnóstico en etapa escolar (p = 0,045) (Figura 1).
Perfil hormonal
El 83,3% (diez pacientes) presentó deficiencia combinada de hormonas hipofisarias (DCHH) y el 16,7% (dos pacientes) presentó déficit aislado de hormona de crecimiento. Los ejes más afectados fueron: tirotropo y corticotropo en 83,3%, somatotropo en 75%, gonadotrofinas en 41,7% y ADH en 8,3%. Solo un paciente presentó deficiencia hipofisaria completa.
La TSH de cordón fue normal en once pacientes (91,7%). El único valor anormal correspondió a un caso de hipotiroidismo central confirmado por sangre venosa.
Imagenología
Se realizaron estudios de resonancia magnética en 11 pacientes. En 10 de ellos (91%) se observaron anomalías estructurales del eje hipotálamo-hipofisario. Los hallazgos más frecuentes fueron: hipófisis hipoplásica o ausente y neurohipófisis ectópica (58,3%), tallo hipofisario ausente o disminuido (50%) y silla turca alterada (33,3%). Solo un paciente presentó imagenología normal a pesar de tener deficiencia hormonal múltiple (Figura 2).
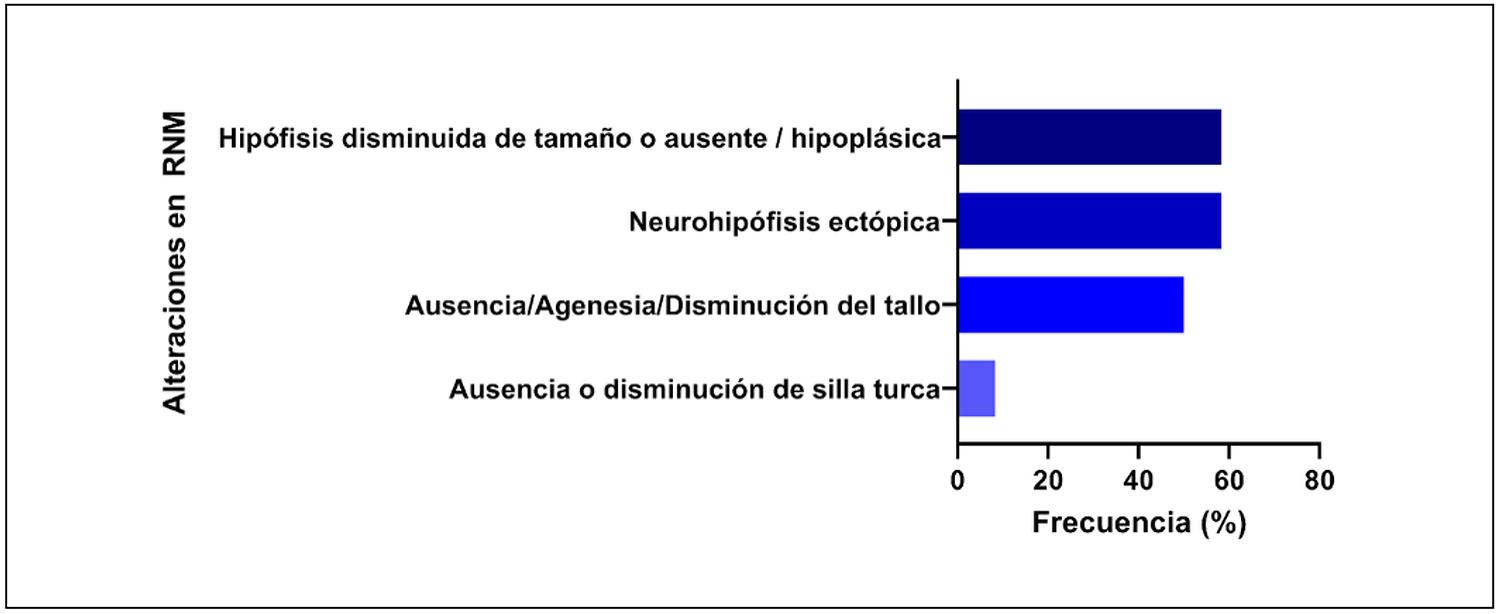
Figura 2. Alteraciones estructurales en la resonancia magnética de pacientes con hipopituitarismo congénito (n = 11).Las anomalías más frecuentes fueron neurohipófisis ectópica (58,3%), tallo hipofisario ausente o disminuido (50%), hipófisis anterior hipoplásica o ausente (50%) y silla turca disminuida o ausente (33,3%).
En tres pacientes se diagnosticó el síndrome de interrupción del tallo hipofisario. Uno de ellos presentó déficit aislado de GH; los otros dos presentaron deficiencia combinada (GH, ACTH, TSH), sin evaluación completa del eje gonadal.
Presentación neonatal y factores asociados
Cinco pacientes presentaron ictericia neonatal. Tres de ellos tenían afectación de los ejes somatotropo, corticotropo y tirotropo; los otros dos, déficit de ACTH y TSH. La hipoglucemia estuvo presente en el 70% de los pacientes con afectación del eje corticotropo y en el 67% de los que presentaban déficit somatotropo.
Discusión
La mayoría de los pacientes incluidos en esta serie fueron diagnosticados en el período neonatal, lo que concuerda con estudios que reportan alta frecuencia de hipoglucemia e ictericia como manifestaciones iniciales6,22.
Un estudio realizado con 52 niños de 12 países mostró que la hipoglucemia fue el síntoma inicial más frecuente, presente en el 54% de los casos. Por otro lado, encontraron que el 63% de los varones presentaban micropene y criptorquidia23. En los casos con diagnóstico más tardío, la talla baja fue el síntoma predominante, patrón también observado por Lammoglia et al. en una serie chilena11,12.
Predominó la deficiencia combinada de hormonas hipofisarias (83,3%), afectando principalmente los ejes corticotropo, tirotropo y somatotropo, similar a lo reportado en series como la de Dutta et al., en la que el 74% presentó deficiencia múltiple14,24.
Los hallazgos de neuroimagen mostraron una alta frecuencia (91% de los pacientes) de alteraciones estructurales, en particular hipoplasia hipofisaria, neurohipófisis ectópica y tallo hipofisario ausente o interrumpido. Este patrón imagenológico es característico del síndrome de interrupción del tallo hipofisario, el cual se asocia con deficiencia múltiple y forma parte del diagnóstico en un número creciente de pacientes13,14,25,26,28.
La TSH de cordón fue normal en la mayoría de los casos, lo que refuerza la limitación de la pesquisa neonatal actual para detectar hipotiroidismo central. La incorporación de la T4 libre como marcador podría mejorar la sensibilidad diagnóstica21,27.
Una de las principales limitaciones de esta cohorte fue la falta de estudios genéticos, situación común en muchos centros latinoamericanos. Se ha demostrado la correlación entre fenotipo clínico y mutaciones en genes como PROP1 y GH18. La disponibilidad de estas pruebas permitiría no solo una mejor caracterización etiológica, sino también estimar el riesgo de progresión y orientar el consejo genético.
Finalmente, se destaca la necesidad de seguimiento endocrinológico especializado y a largo plazo, dado que algunos déficits hormonales pueden aparecer de forma progresiva y condicionar alteraciones en la pubertad, fertilidad, metabolismo y calidad de vida en la adultez18,29,30.
Conclusiones
La mayoría de los pacientes con hipopituitarismo congénito fue diagnosticada en los primeros seis meses de vida, principalmente por hipoglucemia o ictericia. El déficit hormonal combinado fue la forma predominante, con afectación frecuente de los ejes tirotropo, corticotropo y somatotropo.
La hipoglucemia neonatal se asoció significativamente con el diagnóstico temprano.
Las neuroimágenes revelaron alteraciones estructurales en casi todos los casos, siendo útiles para el diagnóstico y clasificación.
La falta de estudios genéticos constituye una limitación para la caracterización etiológica y el pronóstico a largo plazo.
Se destaca la importancia del seguimiento endocrinológico continuo y multidisciplinario en estos pacientes.

