










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkRevista Médica del Uruguay
versión On-line ISSN 1688-0390
Rev. Méd. Urug. vol.26 no.4 Montevideo dic. 2010
Enfermedad de Von Willebrand adquirida en un linfoma linfoplasmocitario/Macroglobulinemia de Waldenström: reporte de caso
Dres. Isabel Moro*, Carolina Oliver†, Mariana Stevenazzi‡,Cecilia Guillermo§, Silvia Pierri¶, Jorge Decaro††
Hospital de Clínicas, Facultad de Medicina. Universidad de la República. Montevideo, Uruguay
Resumen
La enfermedad de Von Willebrand adquirida es una situación infrecuente que se genera en el contexto de enfermedades autoinmunes, síndromes linfoproliferativos y mieloproliferativos. Se presenta el caso clínico de una paciente portadora de un linfoma linfoplasmocitario con enfermedad de Waldenström, presentándose con un síndrome hemorragíparo y alteraciones de la crasis sanguínea a nivel de la vía intrínseca. Las gammapatías monoclonales como la macroglobulinemia de Waldenström suelen presentarse con dosificaciones de IgM variables, siendo, en el caso que se describe, > 11 g/dl. La clínica puede ser muy proteiforme, afectando varios sistemas, presentando en este caso una complicación por adsorción tumoral: la enfermedad de Von Willebrand adquirida. Se indicó tratamiento quimioterápico en base a talidomida, ciclofosfamida y dexametasona para la enfermedad de base, evolucionando favorablemente, remitiendo el síndrome hemorragíparo con tendencia a la normalización de la crasis, de los valores globulares y plaquetarios en forma mantenida luego de seis series de tratamiento. Otras medidas terapéuticas dirigidas a revertir la coagulopatía, como la plasmaféresis, poseen acción transitoria, no estando exentas de riesgo, y se plantean ante hiperviscosidad aguda manifiesta con riesgo vital. La valoración de la paciente en forma interdisciplinaria permitió el mejor acercamiento diagnóstico y terapéutico.
Palabras clave: ENFERMEDADES DE VON WILLEBRAND.
MACROGLOBULINEMIA DE WALDENSTRÖM.
Keywords: VON WILLEBRAND DISEASES.
WALDENSTROM MACROGLOBULINEMIA.
* Asistente de la Cátedra de Hematología, Facultad de Medicina, Universidad de la República. Uruguay.
† Ex Residente de la Cátedra de Hematología, Facultad de Medicina, Universidad de la República. Uruguay.
‡ Profesora Adjunta de Clínica Médica B, Facultad de Medicina, Universidad de la República. Uruguay.
§ Profesora Adjunta de Laboratorio Clínico, Facultad de Medicina, Universidad de la República. Uruguay.
¶ Ex Profesora Adjunta de la Cátedra de Hematología, Facultad de Medicina, Universidad de la República. Uruguay.
†† Profesor de la Cátedra de Medicina Transfusional, Facultad de Medicina, Universidad de la República. Uruguay.
Correspondencia: Dra. Isabel Moro
Cátedra de Hematología, Hospital de Clínicas. Av. Italia s/n. Montevideo, Uruguay.
Correo electrónico: isamorog@yahoo.com
Recibido: 15/7/10.
Aceptado: 25/10/10.
Introducción
La enfermedad de Von Willebrand adquirida (EvWadq) es un desorden hemorragíparo raro, siendo su prevalencia de 0,04%. La EvW congénita es la coagulopatía hereditaria más frecuente contando con una prevalencia de 1% de la población mundial, no habiendo diferencias tanto en la clínica como en el laboratorio entre ambas. Más de 300 casos de EvWadq han sido publicados desde su descripción en 1968 (1). Se caracteriza por sangrados mucosos por defecto en la actividad del factor de Von Willebrand (FvW) en pacientes sin antecedentes de sangrado. Los linfomas linfoplasmocitarios (LLP), según la nueva clasificación de la Organización Mundial de la Salud, se definen como neoplasias de linfocitos B pequeños, linfoplasmocitos y células plasmáticas, que usualmente involucran la médula ósea (MO) y, a veces, ganglios linfáticos, bazo, y otros órganos. Se asocia frecuentemente a una paraproteína de tipo IgM en cuyo caso se denomina LLP con macroglobulinemia de Waldenström (MW) (2). Otro tipo de linfomas también pueden acompañarse de MW. El 9% de las EvWadq se asocian a MW. Se expondrá a continuación un caso clínico que involucra esta asociación (3).
Caso clínico
Paciente de 77 años, sexo femenino, procedente del interior, con antecedentes personales (AP) de diabetes mellitus no insulino dependiente, hipertensión arterial crónica en tratamiento con régimen hiposódico y enalapril 20 mg/día, derivada de un centro hospitalario por elementos de síndrome hemorragíparo y plaquetopenia. Esta última fue diagnosticada hace tres años, con irregular control, agregando en el último año equimosis en los cuatro miembros y en tronco, espontáneos y ante mínimos traumatismos. Epístaxis reiteradas. Hace seis meses agrega disnea y dolor retroesternal ante el esfuerzo moderado, de segundos de duración que ceden con reposo. No consulta durante estos episodios. Del examen físico se destacaba: paciente obesa, palidez cutáneo-mucosa intensa, equímosis extensas en los cuatro miembros y en dorso. No petequias. No sangrados mucosos en curso. Fondo de ojo: hemorragias retinianas en los cuatro cuadrantes, bilateral. Resto del examen normal. La paciente ingresó a sala de medicina. De la valoración paraclínica se destacó lo siguiente: hemograma: hemoglobina (Hb) 7,3 g/dl, volumen corpuscular medio (VCM) 104 fl, hemoglobina corpuscular media (HCM) 32 pg, plaquetas (PLT) 80.000/mm3, glóbulos blancos (GB) 4.000/mm3, neutrófilos 2.070/mm3, linfocitos 2.190/mm3. La lámina periférica evidenció fenómeno de Rouleaux con fórmula de GB normal confirmando plaquetopenia. Velocidad de eritrosedimentación (VES) 140 mm. Crasis: tasa de protrombina (TP): 71%, tiempo de tromboplastina parcial activada (APTT) 63 segundos, tiempo de trombina (TT) 23 segundos. Uricemia 7,9 mg/dl, proteínas totales: 13,2 g/dl.
El encare clínico inicial fue orientado al estudio de una bicitopenia (anemia y plaquetopenia), es decir insuficiencia medular. El síndrome hemorragíparo de tres años de evolución de tipo equimótico y epistaxis, pudo corresponder tanto a una alteración de la fase vasculoplaquetaria de la hemostasis como de la coagulación. Las alteraciones halladas en el fondo de ojo y en la angiografía sugirieron microangiopatía diabética. Por el fenómeno de Rouleaux y la hiperproteinemia total con insuficiencia medular se planteó por frecuencia mieloma múltiple (MM). El síndrome hemorragíparo equimótico, en ausencia de petequias y con prolongación del APTT y del TT, sugirió coagulopatía adquirida (ausencia de antecedentes). Se realizó diagnóstico presuntivo de EvWadq en MM.
Se solicitó proteinograma electroforético (PEF) que reveló una banda monoclonal en la zona media de gamma-globulinas de 7 g/dl. Por inmunofijación, ésta correspondía a inmunoglobulina M/kappa (IgM), cuya dosificación por nefelometría resultó en 11.900 mg/dl, con descenso de las otras inmunoglobulinas. El mielograma reveló una médula ósea rica con megacariocitos, con infiltración de 72% por linfocitos y linfoplasmocitos y 22% de plasmocitos, con escaso remanente mielo-eritroide. El inmunofenotipo (IF) de MO destacó la presencia de 45% de linfocitos B, CD19+, con restricción clonal de la cadena liviana Kappa, débil en membrana y citoplasmática intensa. Esa población expresa CD38+, CD45++ y CD 20+, y no expresan CD 10 ni CD5. Los hallazgos son típicos de LLP. Se concluye el diagnóstico de LLP. No se realiza biopsia de MO dadas las alteraciones coagulopáticas. La función renal estaba levemente alterada: azoemia 53 mg/dl, creatinina 1,40 mg/dl, clearance de creatinina 44 ml/min. Radiografías de esqueleto: sin alteraciones relevantes. La tomografía de cuello, tórax, abdomen y pelvis no detecta adenomegalias ni otras alteraciones. Por presentar un APTT prolongado se realiza mezcla con pool de plasma normal con relación 1:1, corrigiendo el APTT, con un índice de Rossner de 10. Con este resultado se sospechó la carencia de factores de la coagulación, por lo que se dosificó el factor VIII: 14,3%. Seguidamente se dosificó el FvW antigénico: 23,5% y FvW/cofactor ristocetina: 46%. Se confirmó, por lo tanto, el diagnóstico de EvW. En ausencia de antecedentes de sangrados en una paciente de edad avanzada se trataría de una EvWadq. De los hallazgos del mielograma, apoyados por el IF analizado, y las características de la paraproteína se realizó el diagnóstico de LLP con MW(2). Morel y colaboradores desarrollaron un sistema de estadificación pronóstica internacional en MW (ISSMW) en una serie de 587 pacientes. Identificaron la edad > 65 años, Hb < de 11,5 g/dL, nivel de plaquetas < de 100.000x109/l, B2 microglobulina > 3 mg/L y pico monoclonal en sangre > 7 g/dL como factores de mal pronóstico. Los pacientes de bajo riesgo (sin elementos adversos o uno distinto de la edad) tienen una sobrevida media a cinco años de 87%, los de riesgo intermedio (edad > 65 años o 2 factores) 68%, y los de alto riesgo (> 2 factores) 35% (4). Esta paciente posee cinco factores de pronóstico adverso. Se concluye finalmente el diagnóstico de LLP con MW (LLP/MW), asociado a EvWadq, con pronóstico desfavorable.
Tratamiento
La paciente fue valorada en conjunto con la cátedra de medicina transfusional para optimizar el manejo de su coagulopatía. Se optó por una actitud expectante a este respecto dada la ausencia de sangrados de riesgo vital y se consideró que el mejor tratamiento de su coagulopatía sería el tratamiento de la enfermedad de base. Se consideraron como eventualidades el uso de plasmaféresis y concentrados de factor VIII/FvW en caso de aparición de signos de hiperviscosidad o aumento del sangrado. En cuanto a la enfermedad de base (LLP/MW) en una paciente de 77 años se decidió iniciar clorambucil 20 mg/día y prednisona 60 mg/día, ambos en concomitancia por siete días. De la evolución inicial no se evidenció mejoría significativa, por lo cual se cambió el tratamiento a talidomida 100 mg vía oral en forma diaria por seis meses, ciclofosfamida 1 g intravenosa dosis única el día 1, y dexametasona 40 mg intravenosa día por días 1 al 5. La ciclofosfamida y dexametasona fueron cicladas cada 28 días (5-7). La respuesta clínica fue satisfactoria, como se observa en la tabla 1.
Discusión
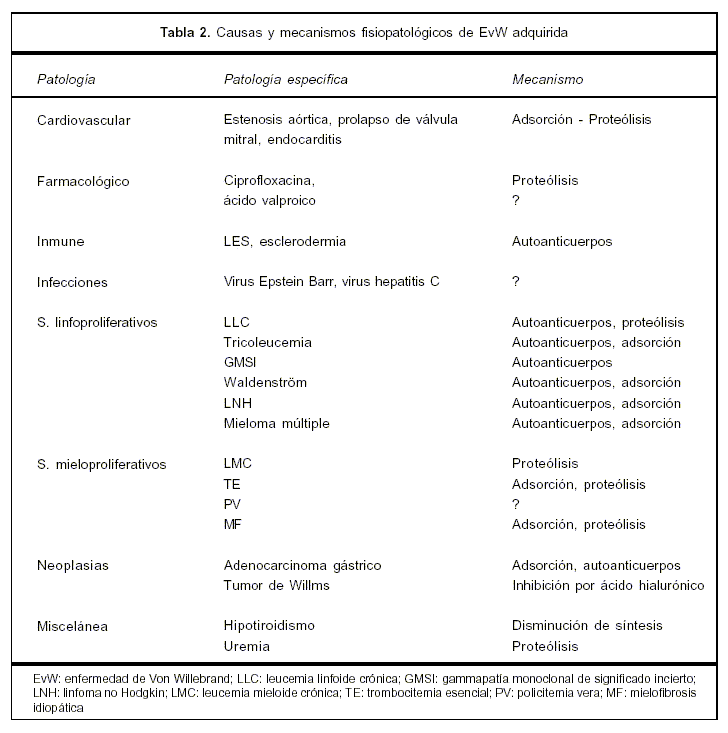
La EvWadq puede desarrollarse en el contexto de numerosas patologías y secundaria a fármacos. Por orden de frecuencia se dividen en seis categorías: síndromes linfoproliferativos (48%), síndromes mieloproliferativos (15%), tumores sólidos (5%), enfermedades autoinmunes (2%), cardiopatías (21%), entre otras causas (28%)(1). La EvWadq, tal como la forma congénita, cursa con un defecto cuanti o cualitativo del FvW, una gran glicoproteína adhesiva que interviene en la hemostasia primaria, actuando en la adhesión de las plaquetas al vaso sanguíneo. Es sintetizado por las células endoteliales y megacariocitos, hallándose en plasma en forma de dímeros de 300 kd, hasta multímeros de 20.000 kd. Estos últimos son los más eficientes para la adhesión plaquetaria(7). Ésta se produce mediante la interacción colágeno-plaqueta a través de los receptores glucoproteína Ib y también la glucoproteína IIbIIIa de las plaquetas, y sitios aminoacídicos bien identificados en el FvW y a través de su dominio A1 con el colágeno. Fisiológicamente se forma una monocapa de plaquetas sobre el subendotelio, que a su vez se une al FvW sucediéndose la agregación plaquetaria. Otra gran función del FvW es su acción estabilizadora del FVIII, el cual al formar un complejo puede protegerse de los inhibidores fisiológicos como la proteína C activada. El descenso del FvW determina consecuentemente el descenso del FVIII (8). El diagnóstico de la EvWadq es esencial, dado el riesgo hemorrágico potencial. Existen tres grandes categorías de EvW: tipo I: déficit cuantitativo parcial del FvW; tipo II: déficit cualitativo, y tipo III: déficit total. En el caso descripto, se trataría de una EvWadq de tipo I (6,9,10). En la forma adquirida de la EvW, la producción y liberación del FvW es normal o aumentada, el mismo disminuye por remoción incrementada por diferentes mecanismos fisiopatológicos: alteración inmunológica, proteólisis, adsorción del FvW por las células tumorales, entre otros(7)(tabla 2).
La causa principal hallada en síndromes linfoproliferativos es la presencia de anticuerpos, pero estos sólo se pueden confirmar en 20% de los casos debido a técnicas de baja sensibilidad y especificidad(7). Se explica por la presencia de un anticuerpo anti FvW contra dominios funcionales (Gp IIB IIIA, IB), determinando disminución de su actividad. Estos son de tipo IgG, raramente IgM, y excepcionalmente IgA(8-10). La presencia de estos anticuerpos se demostró mediante el agregado de plasma de pacientes con EvWadq a plasma normal, mostrando inhibición de la actividad del FvW (CoR)(10). Estos no se hallaron en nuestro caso. Otra causa involucrada en linfomas es la adsorción del FvW por células malignas. Mediante la expresión de moléculas de adhesión aberrantes en la superficie de las células tumorales se produce la unión del FvW a las mismas, lo que determina su disminución en plasma (7,9,10-12). En nuestro caso no se demostró un inhibidor, y el corregir el APTT con pool de plasma normal e in vivo con el tratamiento quimioterápico supone un mecanismo de adsorción por las células tumorales. En la tabla 3 se puede observar la sensibilidad de las pruebas de laboratorio en EvW.
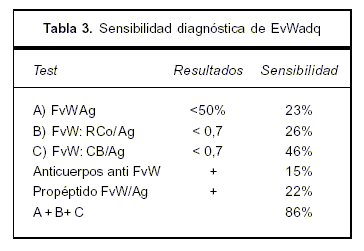
Un número sustancial de casos se presenta con resultados normales de estas pruebas de laboratorio en los cuales es de gran utilidad contar con el estudio molecular de los multímeros si persiste la sospecha clínica(7,9). En algunos casos se puede observar una expresión aberrante de GP IB (normalmente presente en la membrana plaquetaria) por los linfocitos anómalos, objetivable por citometría de flujo(11). El tratamiento de la EvWadq se basa en el tratamiento de la enfermedad de base. Dado que la paciente no presentaba sangrado agudo de riesgo vital, ni se planificaban procedimientos invasivos, no se realizó la terapéutica con desmopresina o concentrados como FVIII/FvW de pureza intermedia o alta (13,14). En caso de no ser efectivo este tratamiento de reposición con factores, se puede utilizar el factor VII activado (14,15). La plasmaféresis no ha demostrado ser muy efectiva para el tratamiento de EvW asociada a linfomas, en cambio el recambio plasmático es indicación categoría I cuando se asocia a la MW con síndrome de hiperviscosidad o crioglobulinemia, o ambos, no presentes en nuestro caso (categoría I ASFA: American Society for Apheresis)(13). El LLP/MW es un linfoma de baja incidencia, de 1 a 6 casos por millón de habitantes con una mediana de edad de presentación de 60 a 71 años, y sobrevida media de cinco años. Constituye menos de 2% de todas las hemopatías malignas y 1,4% de todos los LNH B. Se trata de un linfoma de linfocitos pequeños, linfoplasmocitos y plasmocitos que comprometen MO, ganglios linfáticos y el bazo (15% a 30%) de curso indolente. Puede acompañarse de una paraproteinemia usualmente IgM de cualquier cuantía, a lo que se le denomina MW, como presentó la paciente descripta. Existe una predisposición familiar en 20% de casos. El virus de la hepatitis C ha sido asociado con crioglobulinemia tipo II y LLP en algunas series. Estas pueden ser no progresivas y remitir con el tratamiento antiviral o evolucionar como un linfoma indolente. El motivo de consulta más frecuente es la anemia(2). Los síntomas y signos en LLP/MW pueden ser muy proteiformes. Se agrupan en la tabla 4.
La presencia de un pico IgM no es patognomónico de MW, dado que pueden presentarse acompañando otros linfomas no LLP. También puede preceder al desarrollo de un LLP manifiesto. Las pruebas diagnósticas incluyen el estudio completo de la médula ósea, con inmunofenotipo y biopsia. Para evaluar la extensión lesional se realiza tomografía axial computada (TAC) desde cuello a pelvis para pesquisar la presencia de adenomegalias profundas(2). El tratamiento debe adecuarse a los factores pronósticos y comorbilidades. El propósito de éste es mejorar la calidad de vida con mínimos efectos adversos. Desde el 3º Workshop de MW del año 2006, se ha enfatizado en que el tratamiento debe reservarse para pacientes sintomáticos y no debe tratarse el nivel de IgM sérica. Se consideran los siguientes factores para iniciar el tratamiento: Hb < 10 g/dL, plaquetas < 100.000 x10 9/l, adenomegalias bulky, visceromegalias, hiperviscosidad sintomática, neuropatía, amiloidosis, crioglobulinemia o evidencia de transformación de la enfermedad(4-6). En los casos con síndrome de hiperviscosidad manifiesta es necesaria la realización de plasmaféresis, ya que 80% de la IgM es intravascular(14). Los fármacos de primera línea de tratamiento son el rituximab como monoterapia o asociado a ciclofosfamida, análogos nucleosídicos, bortezomib o talidomida (5,6,15-20). La paciente recibió un tratamiento conservador (edad y comorbilidades), no siendo candidata a tratamientos de mayor intensidad. Actualmente, altas dosis de quimioterapia y trasplante autólogo de progenitores hemopoyéticos se utilizan como estrategia de rescate ante enfermedad refractaria o recaída; el trasplante alogénico sólo en el contexto de ensayos clínicos(5). Esta paciente presenta criterios de tratamiento como: pico monoclonal a IgM > 7 g/dL, citopenias, y como complicación, la EvWadq. Dada la escasa respuesta al clorambucil, se decidió cambiar de plan QT a dexametasona-ciclofosfamida-talidomida, dadas las altas tasas de respuesta en los trabajos que utilizan este plan asociado a rituximab. La evolución fue satisfactoria luego de seis ciclos de quimioterapia (tabla 1).
Conclusiones
Se ha descripto una paciente portadora de un LLP/MW que se presentó clínicamente con un síndrome hemorragíparo catalogado como EvWadq por mecanismo de adsorción tumoral, siendo ambas patologías de muy baja incidencia. El encare interdisciplinario de estos pacientes es fundamental para definir la mejor estrategia terapéutica en forma individualizada, adecuando la intensidad del tratamiento según comorbilidades, factores pronósticos y presencia de complicaciones.
Summary
Acquired Von Willebrand disease is an unusual situation arising within the context of self-immune diseases, lymphoproliferative and mieloproliferative disorders. The study presents the clinical case of a patient carrier of a lymphoplasmocitary lymphoma with Waldenström's disease which presented with a hemorrhagic syndrome and alterations of the blood crasis in the intrinsic way. Monoclonal gammopathies such as Waldenström's macroglobulinemia usually appear with varied IgM dosifications, being it > 11 g/dl in the case described. Clinical symptoms may be very proteiform, affecting several systems, and in this case it presented complications resulting from tumor adsorption: acquired Von Willebrand disease. Chemotherapy with thalidomide, cyclophosphamide and dexamethasone was indicated for the base disease and there was favourable evolution, the hemorrhagic syndrome remitted and there was a tendency to crasis normalization, the same as globular and platelets values ongoing normalization after six series of treatment. Other therapeutic measures aiming to reverse coagulopathy, such as plasmapheresis, are short-acting, and they are not exempt from risks, and they are considered as options upon obvious acute hyperviscosity with a risk of life. Multidisciplinary patient assessment enabled the best diagnosis and therapeutic approach.
Résumé
La maladie de Von Willebrand acquise est une situation peu fréquente qui se génère dans le contexte de maladies auto-immunes, de syndromes lympho-prolifératifs et myélo-prolifératifs. On présente le cas clinique d'une patiente porteuse d'un lymphome lymphoplasmocytaire, qui avait la maladie de Waldenström, qui se présentait avec un syndrome hémorragipare et des altérations de la crase sanguine au niveau de la voie intrinsèque. Les gammapathies monoclonales telles que la macroglobulinémie de Waldenström se présentent d'habitude avec des doses d'IgM variables; dans le cas décri, > 11 g/dl. La clinique peut être très protéiforme, ce qui altère plusieurs systèmes, et a présenté dans ce cas une complication par absorption tumorale : la maladie de Von Willebrand acquise. On a ordonné un traitement de chimiothérapie à base de thalidomide, cyclophosphamide et dexaméthasone pour la maladie de base. La malade a évolué favorablement, le syndrome hémorragipare a reculé et tendait à la normalisation de la crase, des valeurs globulaires et plaquettaires de façon soutenue après six séries de traitement. D'autres mesures thérapeutiques dirigées à renverser la coagulopathie, telle la plasmaphérèse, possèdent une action transitoire, non sans risques, et sont proposées face à l'hyperviscosité aiguë manifeste avec risque de vie. L'évaluation de la patiente de façon interdisciplinaire a permis la meilleure approche diagnostique et thérapeutique.
Resumo
A doença de Von Willebrand adquirida é pouco freqüente e surge no contexto de doenças auto-imunes, síndromes linfoproliferativos e mieloproliferativos. Descrevemos o caso de uma paciente portadora de linfoma linfoplasmocitario com doença de Waldenström, que apresentava uma síndrome hemorragípara e alterações da crase sanguínea da via intrínseca. As gamapatias monoclonais como a macroglobulinemia de Waldenström podem apresentar-se com dosificação variável da IgM que neste caso era > 11 g/dl. O quadro clínico pode ser proteiforme afetando vários sistemas, apresentado nesta paciente uma complicação por adsorção tumoral: a doença de Von Willebrand adquirida. Para o tratamento da doença de base foi indicada quimioterapia com talidomida, ciclofosfamida e dexametasona que levou a uma evolução favorável com remissão da síndrome hemorragípara com tendência à normalização da crase e dos valores de glóbulos e plaquetas depois de seis séries de tratamento. Outras medidas terapêuticas orientadas à reversão da coagulopatia como a plasmaferese possuem ação transitória e não estão isentas de risco e são sugeridas quando há hiperviscosidade aguda com risco vital. A avaliação multidisciplinar da paciente favoreceu o diagnóstico e o tratamento.
Bibliografía
1. Federici AB, Budde U, Rand JH. Acquired von Willebrand syndrome: International Registry diagnosis and management from online to bedside. Hämostaseologie 2004; 24(1): 50-5.
2. Swerdlow SH, Berger F. Lymphoplasmacytic lymphoma: B lymphomas. In: International Agency for Research on Cancer. WHO classification of tumors of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC Press, 2007: 194-5.
3. Federici AB. Acquired von Willebrand syndrome: an underdiagnosed and misdiagnosed bleeding complication in patients with lymphoproliferative and myeloproliferative disorders. Semin Hematol 2006; 43(1 Suppl 1): S48-58.
4. Morel P, Duhamel A, Gobbi P, Dimopoulos MA, Dhodapkar MV, McCoy J, et al. International prognostic scoring system for Waldenstrom macroglobulinemia. Blood 2009; 113(18): 4163-70.
5. Dimopoulos MA, Gertz MA, Kastritis E, Garcia-Sanz R, Kimby EK, Leblond V, et al. Update on treatment recommendations from the Fourth International Workshop on Waldenstrom's Macroglobulinemia. J Clin Oncol 2009; 27(1): 120-6.
6. Treon SP, Patterson CJ, Kimby E, Stone MJ. Advances in the biology and treatment of Waldenstrom´s macroglobulinemia: a report from the 5th International Workshop of Waldenström's macroglobulinemia, Stockholm, Sweden. Clin Lymphoma Myeloma 2009; 9(1): 10-15.
7. Vijay A, Gertz MA. Waldenström macroglobulinemia. Blood 2007; 109(12): 5096-103.
8. Kemball-Cook G, McVey, JH, Tuddenham, E. Structure, biology, and genetics of factor VIII. In: Hoffman R, Benz EJ Jr, Shattil SJ (eds). Hematology: basic principles and practice. 4th ed. New York: Churchill Livingstone, 2005: 2148-49.
9. Bustany S, Gautier P, Lequerrec A, Troussard X, Ollivier Y, Borel-Derlon A. Le syndrome de Willebrand acquis: du diagnostic au traitment. Pathol Biol (Paris) 2009; 57(7-8): 536-42.
10. Sucker C, Michiels JJ, Zotz RB. Causes, etiology and diagnosis of acquired von Willebrand disease: a prospective diagnostic workup to establish the most effective therapeutic strategies. Acta Haematol 2009; 121(2-3): 177-82.
11. Kumar S, Pruthi RK, Nichols WL. Acquired von Willebrand disease. Mayo Clin Proc 2002; 77(2): 181-7.
12. Mohri H. Adcquired von Willebrand syndrome: its pathophysiology, laboratory features and management. J Thromb Thrombolysis 2003; 15(3): 141-9.
13. Szczepiorkowski ZM, Shaz BH, Bandarenko N, Winters JL. The new approach to assignment of ASFA categories introduction to the fourth special issue: clinical applications of therapeutic apheresis. J Clin Apher 2007; 22(3): 96-105.
14. Federici AB, Rand JH, Mannucci PM. Acquired von Willebrand syndrome: an important bleeding complication to be considered in patients with lymphoproliferative and myeloproliferative disorders. Hematol J 2001; 2(6): 358-62.
15. Friederich PW, Wever PC, Briët E, Doorenbos CJ, Levi M. Successful treatment with recombinant factor VIIa of therapy-resistant severe bleeding in a patient with acquired von Willebrand disease. Am J Hematol 2001; 66(4): 292-4.
16. Dimopoulos MA, Merlini G, Leblond V, Anagnostopoulos A, Alexanian R. How we treat Waldenström's macroglobulinemia. Haematologica 2005; 90(1): 117-25.
17. Treon SP. How I treat Waldenström's macroglobulinemia. Blood 2009; 114(12): 2375-85.
18. Leleu X, Gay J, Roccaro AM, Moreau AS, Poulain S, Dulery R, et al. Update on therapeutic options in Waldenström macroglobulinemia. Eur J Hematol 2008; 82(1): 1-12.
19. Mant MJ, Hirsh J, Gauldie J, Bienenstock J, Pineo GF, Luke KH. Von Willebrand's syndrome presenting as an acquired bleeding disorder in association with a monoclonal gammopathy. Blood 1973; 42(3): 429-36.
20. Federici AB, Stabile F, Castaman G, Canciani MT, Mannucci PM. Treatment of acquired von Willebrand syndrome in patients with monoclonal gammopathy of uncertain significance: comparison of three different therapeutic approaches. Blood 1998; 92(8): 2707-11.


{kind=link}
{kind=link}
{kind=link}