










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Links relacionados
Compartir

 Permalink
PermalinkRevista Médica del Uruguay
versión On-line ISSN 1688-0390
Rev. Méd. Urug. vol.31 no.3 Montevideo set. 2015
Linfoma tiroideo, un dilema diagnóstico
María Rosa Finozzi*, Pablo Orellano†, María del Pilar Serra‡
Cátedra de Endocrinología y Metabolismo, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Montevideo, Uruguay
Resumen
Dadas las implicancias diagnóstico-terapéuticas de una tumoración de rápido crecimiento en tiroides, esta situación debe hacer pensar en cáncer indiferenciado y linfoma tiroideo (LT), entidad de baja frecuencia que representa un desafío diagnóstico para el equipo médico. Los más frecuentes son los linfomas no Hodking tipo B (LNHB). La alta tasa de proliferación celular los hace sensibles al tratamiento con poliquimioterapia (PQT). Objetivo: se presenta el caso clínico de un LT primario cuya clasificación inmunohistoquímica (IHQ) es cuestionable. Conclusión: el LT es inusual, se descartará frente a una masa tiroidea, de rápido crecimiento, planteándolo siempre como diagnóstico diferencial. El tratamiento debe realizarse con equipo multidisciplinario, en base a PQT inicial y radioterapia, siendo la cirugía una terapéutica controversial.
Palabras clave: LINFOMA NEOPLASIAS DE LA TIROIDES
Key words: LYMPHOMA THYROID NEOPLASMS
* Asistente de la Clínica de Endocrinología y Metabolismo, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay.
† Prof. Adj. Clínica de Endocrinología y Metabolismo, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay.
‡ Prof. Agregada Clínica de Endocrinología y Metabolismo, Hospital de Clínicas, Facultad de Medicina, Universidad de la República. Uruguay.
Correspondencia: Dra. Rosa Finozzi. Dirección: Av. Italia s/n, Hospital de Clínicas, Cátedra de Endocrinología. Correo electrónico: rofinozzi@gmail.com
Recibido: 8/12/14 Aceptado: 20/7/15
Introducción
La mayoría de los linfomas tiroideos son de tipo no Hodking, siendo los linfomas Hodking muy raros(1).
Presentan una incidencia anual de 2 por millón(2,3). Solo el 2% de los linfomas extranodales se originan dentro de la glándula tiroides, representando no más de 2% de todos los tumores tiroideos(2).
Los pacientes suelen presentarse con una masa tiroidea de rápido crecimiento asociada a síntomas locorregionales como disfonía, estridor o disfagia, acompañado de síntomas B, como ser fiebre mayor a 38 ºC, sudores nocturnos, pérdida de peso mayor a 10% del peso normal en un período de seis meses o menos, y prurito(4).
Caso clínico
Paciente de sexo masculino de 59 años, oligofrénico, que consulta por una masa tiroidea pétrea de 8 cm de diámetro mayor, de crecimiento rápido con elementos compresivos locorregionales como disfagia y disfonía. Funcionalmente presenta hipotiroidismo clínico severo con serositis pleural y pericárdica asociado a neumopatía aguda comunitaria (NAC) en hemitórax izquierdo. Sin síntomas B.
De la analítica inicial destacamos TSH >60 mUI/L (VN 0,27-4,6), T4L <0,46 ng/dL (0,7-1,2) confirmando el diagnóstico clínico de hipotiroidismo primario.
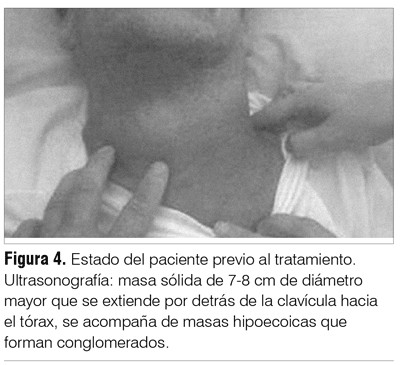
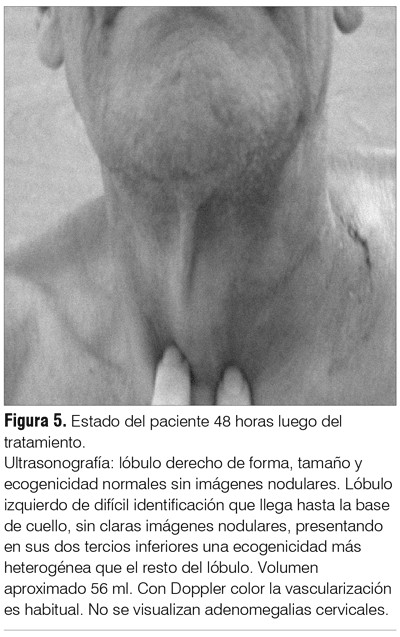
La ultrasonografía de cuello pretratamiento informó una masa sólida de 7-8 cm de diámetro mayor que se extiende por detrás de la clavícula hacia el tórax, acompañada de masas hipoecoicas formando conglomerados.
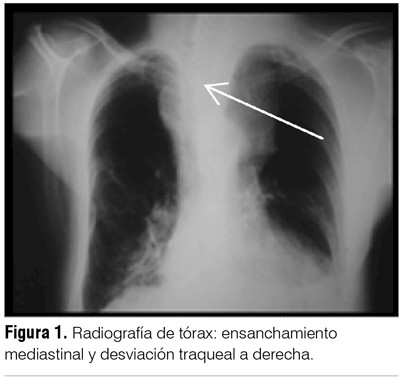
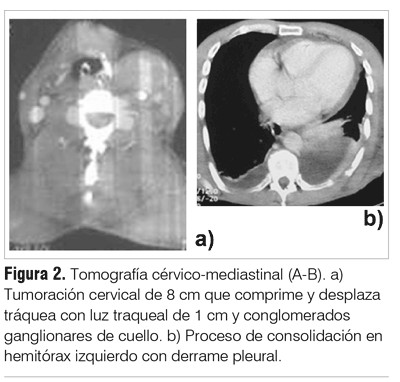
La radiografía de tórax mostró ensanchamiento mediastinal y desviación traqueal a derecha (figura 1). La tomografía cérvico-mediastinal evidenció tumoración cervical de 8 cm que comprimía y desplazaba tráquea, con luz traqueal de 1 cm y conglomerados ganglionares de cuello (figura 2 A). Proceso de consolidación en hemitórax izquierdo con derrame pleural (DP) (figura 2 B).
Por presentar síntomas cardiovasculares se solicitó ecocardiograma que mostró un leve derrame pericárdico a predominio de sector anterior y derecho, sin elementos de taponamiento y derrame pleural izquierdo.
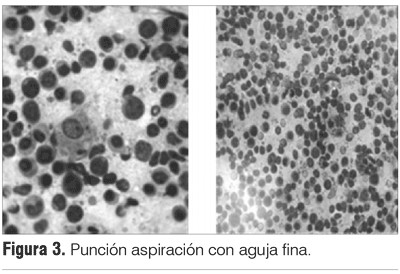
Por la masa de rápido crecimiento se planteó como diagnóstico clínico presuntivo el carcinoma anaplásico o el linfoma tiroideo. Se realizó una punción aspiración con aguja fina (PAAF) para análisis citológico, siendo concluyente con linfoma no Hodking (LNH) (figura 3).
Continuando con el diagnóstico paraclínico se procedió a realizar la biopsia del conglomerado ganglionar con inmunofenotipo por citometría de flujo e IHQ, realizándose panel de combinaciones múltiples en las que se han incluido los marcadores para: CD45, CD3, CD4, CD8, CD19, CD56, CD20, CD10, BCL2, CD34, CD38, CD2, CD5, CD23 y para cadenas livianas (K/L). Las células recuperadas de la biopsia ganglionar tienen características linfoides en el 12% del total de las células nucleadas, y, de éstas, los elementos de la línea B (CD19) representan el 51,3%, mientras que los elementos de línea T son el 46,1%.
Los elementos de la línea B no presentan patrón clonal con las cadenas livianas. Los elementos de la línea T se subdividen en 25,7% de T cooperadores (CD3+, CD4-, CD8-), 64,8% de T supresores (CD 3+, CD4+, CD8-), 3,6% de T doble positivo (CD3, CD4, CD8+) y 5% de T doble negativos (CD3+, CD4-CD8). Existen, además, 7,4% de PMN maduros y células que no se marcaron con los anticuerpos monoclonales empleados (80%). Se identificaron elementos linfoides y PMN.
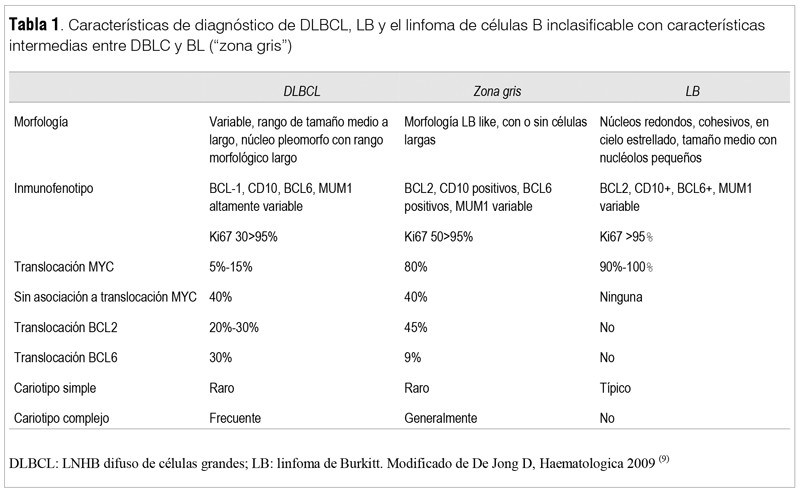
El hallazgo del 80% de células que no se identifican con las líneas hematopoyéticas plantea la posibilidad de una sustitución por proliferación extrahematopoyética. La anatomía patológica informa presencia de necrosis extensa del parénquima ganglionar con áreas de infarto, observándose en el único sector remanente una proliferación celular de patrón sólido con aspecto de “cielo estrellado”, con macrófagos, apoptosis y frecuentes figuras de mitosis. Células de mediana y pequeña talla, núcleo oval, con membrana nuclear evidente y células aisladas de mayor talla. El inmunofenotipo identificó elementos linfoides y polimorfonucleares sin patrón clonal y la IHQ informó: CK-, ACL+, CD20+, CD10+ focal, Ki67 índice de proliferación >70%, CD15-, CD30-, Bcl2-, CD3-, tiroglobulina-. En suma: linfoma no Hodking de células B (LNHB), con alto índice de proliferación, superior al 70%, inmunomarcación KI67, lo cual es sugestivo de un linfoma de Burkitt. No pudiendo descartarse linfoma de Hodking inclasificable o linfoma B de alto grado inmunoblástico, polimorfo inclasificable, con figuras intermedias entre LNHB difuso de células grandes (DLBCL) y linfoma de Burkitt. Queda inconcluso el inmunofenotipo, siendo necesario para un correcto diagnóstico el hallazgo del protooncogén C-MYC (criterio diagnóstico definitivo), estudio con el que no se pudo contar en nuestro paciente.
Dado el diagnóstico confirmatorio de hipotiroidismo primario y linfoma tiroideo asociado a una NAC, se inició tratamiento con levotiroxina en dosis progresivas, antibioticoterapia y dos ciclos de poliquimioterapia con ciclofosfamida, adriamicina, vincristina y prednisona. Presentó una excelente respuesta en lo inmediato en lo funcional, estructural e infeccioso, quedando pendiente el estudio molecular del protooncogén C-MYC del que se esperaba determinar el subtipo de LNHB, lo que hubiera permitido establecer un pronóstico y tratamiento definitivo.
Desconocemos la evolución posterior dado que el paciente, una vez dado de alta, no regresó a los controles.
En las figuras 4 y 5 mostramos al paciente pre y postratamiento junto con la ultrasonografía comparativa.
Discusión
EL LT primario es conocido con una incidencia menor al 2% de los cánceres tiroideos(2). Es más frecuente en el sexo femenino (3:1)(5,6), con una edad media entre 60-75 años(7). De los mismos, 10% a 20% presentan hipotiroidismo asociado a una tiroiditis de Hashimoto (TH). La existencia previa de una tiroiditis crónica autoinmune TH es un factor de riesgo conocido para linfoma, presentándose en la mitad de los pacientes. Su antecedente aumenta el riesgo de tener un linfoma 60 veces más que los pacientes sin tiroiditis(5,6,8). En nuestro paciente no contamos con el resultado del análisis de los anticuerpos antitiroideos para realizar el diagnóstico de TH.
Su estirpe celular pertenece a los LNHB, siendo extremadamente raros los de estirpe T o los LH. Sin embargo, la tipificación del tipo B presenta controversias, como sucede en este paciente.
La dificultad se centró en diferenciar el tipo de linfoma B de alto grado del LB, ya que tiene connotaciones terapéuticas y pronósticas.
EL LB está constituido por una proliferación clonal de linfocitos pobremente diferenciados de estirpe B de rápida replicación que expresa el protooncogén C-MYC (criterio diagnóstico definitivo); estudio con el que no contamos en nuestro medio (tabla 1)(9).
Pocos casos confinados solo a la tiroides se describen en la literatura para este tipo de LNH agresivo.
No fue posible descartar la posibilidad de estar frente a un LNHB de características intermedias entre los DLBCL y el LB con un pronóstico más favorable, requiriendo para su diagnóstico un riguroso análisis de las características histológicas, inmunofenotípicas y genéticas(9).
Conclusiones
El LT es inusual, se debe pensar en este diagnóstico nosológico frente a una masa tiroidea, de rápido crecimiento, fundamentalmente en pacientes con diagnóstico de enfermedad de Hashimoto previa, que en determinado momento y a pesar de tratamiento con levotiroxina, el bocio aumenta su tamaño, o en casos como el presente, en que sin diagnóstico previo de TH se asiste a un aumento progresivo de la tiroides, orientador de neoplasia de este órgano. Por razones de frecuencia, el diagnóstico de LT es de descarte(10).
El tratamiento debe realizarse con equipo multidisciplinario, en base a PQT inicial y radioterapia, siendo la cirugía una terapéutica controversial(11).
Abstract
Given diagnostic-therapeutic implications of a fast growing tumor in the thyroid, it is natural to think of undifferentiated cancer and thyroid lymphoma, a low frequency entity that entails a diagnostic challenge for the medical team. Type B non-Hodking lymphomas (NHL) are the most frequent ones. The high rate of cell proliferation makes them sensitive to polychemotherapy treatment.
Objective: the clinical case of a thyroid lymphoma of a questionable immunohistochemical classification is presented.
Conclusion: thyroid lymphomas are unusual, and they will be ruled out upon a fast growing thyroid mass, always considering it as a differential diagnosis.
A multidisciplinary team must be involved in the treatment, based on initial polychemotherapy and radiotherapy, surgery being a controversial therapy.
Resumo
Considerando as implicações diagnóstico-terapêuticas de um tumor de tireoides que cresce rapidamente, deve-se pensar em câncer indiferenciado e linfoma da tireoide (LT), uma entidade pouco frequente cujo diagnóstico desafia os médicos. Os mais frequentes são os Linfomas não Hodgkin tipo B (LNHB). Sua alta taxa de proliferação celular faz com que sejam sensíveis ao tratamento com poliquimioterapia (PQT).
Objetivo: apresenta-se o caso clínico de um LT primário cuja classificação imuno-histoquímica (IHQ) foi questionada.
Conclusão: o LT é raro e deve ser descartado quando se observa uma massa tireóidea, de rápido crescimento, considerando-o sempre como diagnóstico diferencial.
O tratamento deve ser realizado por uma equipe multiprofissional empregando PQT no início e radioterapia, sendo a cirurgia uma terapêutica controversa.
Bibliografía
1. Wang SA, Rahemtullah A, Faquin WC, Roepke J, Harris NL, Hasserjian RP. Hodgkin’s lymphoma of the thyroid: a clinicopathologic study of five cases and review of the literature. Mod Pathol 2005; 18(12):1577-84.
2. Freeman C, Berg JW, Cutler SJ. Occurrence and prognosis of extranodal lymphomas. Cancer 1972; 29(1):252-60.
3. Graff-Baker A, Sosa JA, Roman SA. Primary thyroid lymphoma: a review of recent developments in diagnosis and histology-driven treatment. Curr Opin Oncol 2010; 22(1):17-22.
4. Agarwaf N, Wangnoo SK, Sidiqqi A, Gupt M. Primary thyroid lymphoma: a series of two cases and review of literature. J Assoc Physicians India 2013; 61(7):496-8.
5. Pedersen RK, Pedersen NT. Primary non-Hodgkin’s lymphoma of the thyroid gland: a population based study. Histopathology 1996; 28(1):25-32.
6. Holm LE, Blomgren H, Löwhagen T. Cancer risks in patients with chronic lymphocytic thyroiditis. N Engl J Med 1985; 312(10):601-4.
7. Skarsgard ED, Connors JM, Robins RE. A current analysis of primary lymphoma of the thyroid. Arch Surg 1991; 126(10):1199-203.
8. Hyjek E, Isaacson PG. Primary B cell lymphoma of the thyroid and its relationship to Hashimoto’s thyroiditis. Hum Pathol 1988; 19(11):1315-26.
9. de Jong D. Novel lymphoid neoplasms –the borderland between diffuse large B-cell lymphoma and Burkitt’s lymphoma. Haematologica 2009; 94(7):894-6.
10. Gac P, Cabané P, Amat J, Zamorano S, Pineda P, Morales C, et al. Linfoma primario de tiroides: reporte de cuatro casos, Rev Méd Chile 2009; 137(7):928-35.
11. Frizzell JD, Perkins BJ, Morehead RS. Thyroid Lymphoma as a cause of dysphagia and dyspnea in a patient without palpable nodules or Goiter. Case Rep Med 2009; 2009: 385461.


{kind=link}